Jun Hu , Kai-Xin Chen , Bing Rao , Jing-Yuan Ni , Maha A. Thafar , Somayah Albaradei , Muhammad Arif
{"title":"基于蛋白质语言模型和交叉注意力机制的蛋白质-肽结合残基预测","authors":"Jun Hu , Kai-Xin Chen , Bing Rao , Jing-Yuan Ni , Maha A. Thafar , Somayah Albaradei , Muhammad Arif","doi":"10.1016/j.ab.2024.115637","DOIUrl":null,"url":null,"abstract":"<div><p>Accurate identifications of protein-peptide binding residues are essential for protein-peptide interactions and advancing drug discovery. To address this problem, extensive research efforts have been made to design more discriminative feature representations. However, extracting these explicit features usually depend on third-party tools, resulting in low computational efficacy and suffering from low predictive performance. In this study, we design an end-to-end deep learning-based method, E2EPep, for protein-peptide binding residue prediction using protein sequence only. E2EPep first employs and fine-tunes two state-of-the-art pre-trained protein language models that can extract two different high-latent feature representations from protein sequences relevant for protein structures and functions. A novel feature fusion module is then designed in E2EPep to fuse and optimize the above two feature representations of binding residues. In addition, we have also design E2EPep+, which integrates E2EPep and PepBCL models, to improve the prediction performance. Experimental results on two independent testing data sets demonstrate that E2EPep and E2EPep + could achieve the average AUC values of 0.846 and 0.842 while achieving an average Matthew's correlation coefficient value that is significantly higher than that of existing most of sequence-based methods and comparable to that of the state-of-the-art structure-based predictors. Detailed data analysis shows that the primary strength of E2EPep lies in the effectiveness of feature representation using cross-attention mechanism to fuse the embeddings generated by two fine-tuned protein language models. The standalone package of E2EPep and E2EPep + can be obtained at <span><span>https://github.com/ckx259/E2EPep.git</span><svg><path></path></svg></span> for academic use only.</p></div>","PeriodicalId":7830,"journal":{"name":"Analytical biochemistry","volume":"694 ","pages":"Article 115637"},"PeriodicalIF":2.6000,"publicationDate":"2024-08-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Protein-peptide binding residue prediction based on protein language models and cross-attention mechanism\",\"authors\":\"Jun Hu , Kai-Xin Chen , Bing Rao , Jing-Yuan Ni , Maha A. Thafar , Somayah Albaradei , Muhammad Arif\",\"doi\":\"10.1016/j.ab.2024.115637\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Accurate identifications of protein-peptide binding residues are essential for protein-peptide interactions and advancing drug discovery. To address this problem, extensive research efforts have been made to design more discriminative feature representations. However, extracting these explicit features usually depend on third-party tools, resulting in low computational efficacy and suffering from low predictive performance. In this study, we design an end-to-end deep learning-based method, E2EPep, for protein-peptide binding residue prediction using protein sequence only. E2EPep first employs and fine-tunes two state-of-the-art pre-trained protein language models that can extract two different high-latent feature representations from protein sequences relevant for protein structures and functions. A novel feature fusion module is then designed in E2EPep to fuse and optimize the above two feature representations of binding residues. In addition, we have also design E2EPep+, which integrates E2EPep and PepBCL models, to improve the prediction performance. Experimental results on two independent testing data sets demonstrate that E2EPep and E2EPep + could achieve the average AUC values of 0.846 and 0.842 while achieving an average Matthew's correlation coefficient value that is significantly higher than that of existing most of sequence-based methods and comparable to that of the state-of-the-art structure-based predictors. Detailed data analysis shows that the primary strength of E2EPep lies in the effectiveness of feature representation using cross-attention mechanism to fuse the embeddings generated by two fine-tuned protein language models. The standalone package of E2EPep and E2EPep + can be obtained at <span><span>https://github.com/ckx259/E2EPep.git</span><svg><path></path></svg></span> for academic use only.</p></div>\",\"PeriodicalId\":7830,\"journal\":{\"name\":\"Analytical biochemistry\",\"volume\":\"694 \",\"pages\":\"Article 115637\"},\"PeriodicalIF\":2.6000,\"publicationDate\":\"2024-08-08\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Analytical biochemistry\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S0003269724001817\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Analytical biochemistry","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0003269724001817","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
Protein-peptide binding residue prediction based on protein language models and cross-attention mechanism
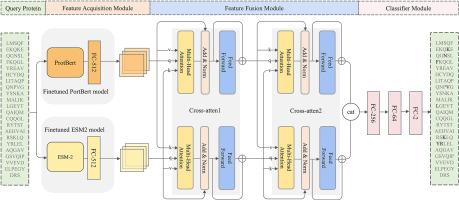
Accurate identifications of protein-peptide binding residues are essential for protein-peptide interactions and advancing drug discovery. To address this problem, extensive research efforts have been made to design more discriminative feature representations. However, extracting these explicit features usually depend on third-party tools, resulting in low computational efficacy and suffering from low predictive performance. In this study, we design an end-to-end deep learning-based method, E2EPep, for protein-peptide binding residue prediction using protein sequence only. E2EPep first employs and fine-tunes two state-of-the-art pre-trained protein language models that can extract two different high-latent feature representations from protein sequences relevant for protein structures and functions. A novel feature fusion module is then designed in E2EPep to fuse and optimize the above two feature representations of binding residues. In addition, we have also design E2EPep+, which integrates E2EPep and PepBCL models, to improve the prediction performance. Experimental results on two independent testing data sets demonstrate that E2EPep and E2EPep + could achieve the average AUC values of 0.846 and 0.842 while achieving an average Matthew's correlation coefficient value that is significantly higher than that of existing most of sequence-based methods and comparable to that of the state-of-the-art structure-based predictors. Detailed data analysis shows that the primary strength of E2EPep lies in the effectiveness of feature representation using cross-attention mechanism to fuse the embeddings generated by two fine-tuned protein language models. The standalone package of E2EPep and E2EPep + can be obtained at https://github.com/ckx259/E2EPep.git for academic use only.
期刊介绍:
The journal''s title Analytical Biochemistry: Methods in the Biological Sciences declares its broad scope: methods for the basic biological sciences that include biochemistry, molecular genetics, cell biology, proteomics, immunology, bioinformatics and wherever the frontiers of research take the field.
The emphasis is on methods from the strictly analytical to the more preparative that would include novel approaches to protein purification as well as improvements in cell and organ culture. The actual techniques are equally inclusive ranging from aptamers to zymology.
The journal has been particularly active in:
-Analytical techniques for biological molecules-
Aptamer selection and utilization-
Biosensors-
Chromatography-
Cloning, sequencing and mutagenesis-
Electrochemical methods-
Electrophoresis-
Enzyme characterization methods-
Immunological approaches-
Mass spectrometry of proteins and nucleic acids-
Metabolomics-
Nano level techniques-
Optical spectroscopy in all its forms.
The journal is reluctant to include most drug and strictly clinical studies as there are more suitable publication platforms for these types of papers.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


