{"title":"美国和欧盟《口服吸入药物产品比较效力和安全性确定指南》之间的区别和相似之处的相关性。","authors":"Gur Jai Pal Singh","doi":"10.1016/j.ejps.2024.106872","DOIUrl":null,"url":null,"abstract":"<div><p>Approval of drug products for market registration warrants, among other data, evidence to support their safety and effectiveness in the target populations. The extent of investigations to provide the supporting evidence varies between the new innovator products and their follow-on versions generally referred to as Generic Drugs Products in the United States and Hybrids in the Europe. The new drug applications entail large data sets encompassing both nonclinical and clinical product developments. Safety and effectiveness in man is studied in sequentially phased clinical trials, including post marketing evaluations (Where applicable). However, for the generic/hybrid products the safety and effectiveness are established through determination of bioequivalence in head-to-head comparison between the originator and the follow-ons. Methods for documentation of bioequivalence for drug products that reach target site(s) through systemic circulation are aligned worldwide. However, establishing bioequivalence of orally inhaled drug products is complex as drug delivery to the local site(s) of action is independent of the systemic circulation. Documentation of bioequivalence gets further complicated due to the Drug-Device combination nature of these products. The guidelines for establishment of BE of locally acting orally inhaled drugs products vary among certain geographies. This article examines the scientific underpinning of distinctions and similarities between the US and EU guidelines.</p></div>","PeriodicalId":12018,"journal":{"name":"European Journal of Pharmaceutical Sciences","volume":"201 ","pages":"Article 106872"},"PeriodicalIF":4.3000,"publicationDate":"2024-08-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S0928098724001842/pdfft?md5=74c03199e7ed5af2bd20fff39f13d123&pid=1-s2.0-S0928098724001842-main.pdf","citationCount":"0","resultStr":"{\"title\":\"Relevance of distinctions and parallels between the US and EU guidelines for determination of comparative effectiveness and safety of the orally inhaled drug products\",\"authors\":\"Gur Jai Pal Singh\",\"doi\":\"10.1016/j.ejps.2024.106872\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Approval of drug products for market registration warrants, among other data, evidence to support their safety and effectiveness in the target populations. The extent of investigations to provide the supporting evidence varies between the new innovator products and their follow-on versions generally referred to as Generic Drugs Products in the United States and Hybrids in the Europe. The new drug applications entail large data sets encompassing both nonclinical and clinical product developments. Safety and effectiveness in man is studied in sequentially phased clinical trials, including post marketing evaluations (Where applicable). However, for the generic/hybrid products the safety and effectiveness are established through determination of bioequivalence in head-to-head comparison between the originator and the follow-ons. Methods for documentation of bioequivalence for drug products that reach target site(s) through systemic circulation are aligned worldwide. However, establishing bioequivalence of orally inhaled drug products is complex as drug delivery to the local site(s) of action is independent of the systemic circulation. Documentation of bioequivalence gets further complicated due to the Drug-Device combination nature of these products. The guidelines for establishment of BE of locally acting orally inhaled drugs products vary among certain geographies. This article examines the scientific underpinning of distinctions and similarities between the US and EU guidelines.</p></div>\",\"PeriodicalId\":12018,\"journal\":{\"name\":\"European Journal of Pharmaceutical Sciences\",\"volume\":\"201 \",\"pages\":\"Article 106872\"},\"PeriodicalIF\":4.3000,\"publicationDate\":\"2024-08-06\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.sciencedirect.com/science/article/pii/S0928098724001842/pdfft?md5=74c03199e7ed5af2bd20fff39f13d123&pid=1-s2.0-S0928098724001842-main.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"European Journal of Pharmaceutical Sciences\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S0928098724001842\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"PHARMACOLOGY & PHARMACY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"European Journal of Pharmaceutical Sciences","FirstCategoryId":"3","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0928098724001842","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"PHARMACOLOGY & PHARMACY","Score":null,"Total":0}
Relevance of distinctions and parallels between the US and EU guidelines for determination of comparative effectiveness and safety of the orally inhaled drug products
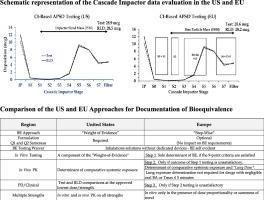
Approval of drug products for market registration warrants, among other data, evidence to support their safety and effectiveness in the target populations. The extent of investigations to provide the supporting evidence varies between the new innovator products and their follow-on versions generally referred to as Generic Drugs Products in the United States and Hybrids in the Europe. The new drug applications entail large data sets encompassing both nonclinical and clinical product developments. Safety and effectiveness in man is studied in sequentially phased clinical trials, including post marketing evaluations (Where applicable). However, for the generic/hybrid products the safety and effectiveness are established through determination of bioequivalence in head-to-head comparison between the originator and the follow-ons. Methods for documentation of bioequivalence for drug products that reach target site(s) through systemic circulation are aligned worldwide. However, establishing bioequivalence of orally inhaled drug products is complex as drug delivery to the local site(s) of action is independent of the systemic circulation. Documentation of bioequivalence gets further complicated due to the Drug-Device combination nature of these products. The guidelines for establishment of BE of locally acting orally inhaled drugs products vary among certain geographies. This article examines the scientific underpinning of distinctions and similarities between the US and EU guidelines.
期刊介绍:
The journal publishes research articles, review articles and scientific commentaries on all aspects of the pharmaceutical sciences with emphasis on conceptual novelty and scientific quality. The Editors welcome articles in this multidisciplinary field, with a focus on topics relevant for drug discovery and development.
More specifically, the Journal publishes reports on medicinal chemistry, pharmacology, drug absorption and metabolism, pharmacokinetics and pharmacodynamics, pharmaceutical and biomedical analysis, drug delivery (including gene delivery), drug targeting, pharmaceutical technology, pharmaceutical biotechnology and clinical drug evaluation. The journal will typically not give priority to manuscripts focusing primarily on organic synthesis, natural products, adaptation of analytical approaches, or discussions pertaining to drug policy making.
Scientific commentaries and review articles are generally by invitation only or by consent of the Editors. Proceedings of scientific meetings may be published as special issues or supplements to the Journal.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


