Vaishnav Krishnan, Jun Wu, Arindam Ghosh Mazumder, Jessica L. Kamen, Catharina Schirmer, Nandani Adhyapak, John Samuel Bass, Samuel C. Lee, Atul Maheshwari, Gemma Molinaro, Jay R. Gibson, Kimberly M. Huber, Berge A. Minassian
{"title":"临床病理分离:马林 KO 小鼠体内的大量拉弗拉体积聚,而家庭笼养小鼠的行为却无明显变化。","authors":"Vaishnav Krishnan, Jun Wu, Arindam Ghosh Mazumder, Jessica L. Kamen, Catharina Schirmer, Nandani Adhyapak, John Samuel Bass, Samuel C. Lee, Atul Maheshwari, Gemma Molinaro, Jay R. Gibson, Kimberly M. Huber, Berge A. Minassian","doi":"10.1002/cne.25660","DOIUrl":null,"url":null,"abstract":"<div>\n \n <p>Lafora disease (LD) is a syndrome of progressive myoclonic epilepsy and cumulative neurocognitive deterioration caused by recessively inherited genetic lesions of EPM2A (laforin) or NHLRC1 (malin). Neuropsychiatric symptomatology in LD is thought to be directly downstream of neuronal and astrocytic polyglucosan aggregates, termed Lafora bodies (LBs), which faithfully accumulate in an age-dependent manner in all mouse models of LD. In this study, we applied home-cage monitoring to examine the extent of neurobehavioral deterioration in a model of malin-deficient LD as a means to identify robust preclinical endpoints that may guide the selection of novel genetic treatments. At 6 weeks, ∼6–7 months, and ∼12 months of age, malin-deficient mice (“KO”) and wild-type (WT) littermates underwent a standardized home-cage behavioral assessment designed to non-obtrusively appraise features of rest/arousal, consumptive behaviors, risk aversion, and voluntary wheel-running. At all timepoints, and over a range of metrics that we report transparently, WT and KO mice were essentially indistinguishable. In contrast, within WT mice compared across the same timepoints, we identified age-related nocturnal hypoactivity, diminished sucrose preference, and reduced wheel-running. Neuropathological examinations in subsets of the same mice revealed expected age-dependent LB accumulation, gliosis, and microglial activation in cortical and subcortical brain regions. At 12 months of age, despite the burden of neocortical LBs, we did not identify spontaneous seizures during an electroencephalographic (EEG) survey, and KO and WT mice exhibited similar spectral EEG features. However, in an in vitro assay of neocortical function, paroxysmal bursts of network activity (UP states) in KO slices were more prolonged at 3 and 6 months of age, but similar to WT at 12 months. KO mice displayed a distinct response to pentylenetetrazole, with a greater incidence of clonic seizures and a more pronounced postictal suppression of movement, feeding, and drinking behavior. Together, these results highlight the clinicopathologic dissociation in a mouse model of LD, where the accrual of LBs may latently modify cortical circuit function and seizure threshold without clinically meaningful changes in home-cage behavior. Our findings allude to a delay between LB accumulation and neurobehavioral decline in LD: one that may provide a window for treatment, and whose precise duration may be difficult to ascertain within the typical lifespan of a laboratory mouse.</p>\n </div>","PeriodicalId":15552,"journal":{"name":"Journal of Comparative Neurology","volume":"532 7","pages":""},"PeriodicalIF":2.1000,"publicationDate":"2024-07-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Clinicopathologic Dissociation: Robust Lafora Body Accumulation in Malin KO Mice Without Observable Changes in Home-Cage Behavior\",\"authors\":\"Vaishnav Krishnan, Jun Wu, Arindam Ghosh Mazumder, Jessica L. Kamen, Catharina Schirmer, Nandani Adhyapak, John Samuel Bass, Samuel C. Lee, Atul Maheshwari, Gemma Molinaro, Jay R. Gibson, Kimberly M. Huber, Berge A. Minassian\",\"doi\":\"10.1002/cne.25660\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div>\\n \\n <p>Lafora disease (LD) is a syndrome of progressive myoclonic epilepsy and cumulative neurocognitive deterioration caused by recessively inherited genetic lesions of EPM2A (laforin) or NHLRC1 (malin). Neuropsychiatric symptomatology in LD is thought to be directly downstream of neuronal and astrocytic polyglucosan aggregates, termed Lafora bodies (LBs), which faithfully accumulate in an age-dependent manner in all mouse models of LD. In this study, we applied home-cage monitoring to examine the extent of neurobehavioral deterioration in a model of malin-deficient LD as a means to identify robust preclinical endpoints that may guide the selection of novel genetic treatments. At 6 weeks, ∼6–7 months, and ∼12 months of age, malin-deficient mice (“KO”) and wild-type (WT) littermates underwent a standardized home-cage behavioral assessment designed to non-obtrusively appraise features of rest/arousal, consumptive behaviors, risk aversion, and voluntary wheel-running. At all timepoints, and over a range of metrics that we report transparently, WT and KO mice were essentially indistinguishable. In contrast, within WT mice compared across the same timepoints, we identified age-related nocturnal hypoactivity, diminished sucrose preference, and reduced wheel-running. Neuropathological examinations in subsets of the same mice revealed expected age-dependent LB accumulation, gliosis, and microglial activation in cortical and subcortical brain regions. At 12 months of age, despite the burden of neocortical LBs, we did not identify spontaneous seizures during an electroencephalographic (EEG) survey, and KO and WT mice exhibited similar spectral EEG features. However, in an in vitro assay of neocortical function, paroxysmal bursts of network activity (UP states) in KO slices were more prolonged at 3 and 6 months of age, but similar to WT at 12 months. KO mice displayed a distinct response to pentylenetetrazole, with a greater incidence of clonic seizures and a more pronounced postictal suppression of movement, feeding, and drinking behavior. Together, these results highlight the clinicopathologic dissociation in a mouse model of LD, where the accrual of LBs may latently modify cortical circuit function and seizure threshold without clinically meaningful changes in home-cage behavior. Our findings allude to a delay between LB accumulation and neurobehavioral decline in LD: one that may provide a window for treatment, and whose precise duration may be difficult to ascertain within the typical lifespan of a laboratory mouse.</p>\\n </div>\",\"PeriodicalId\":15552,\"journal\":{\"name\":\"Journal of Comparative Neurology\",\"volume\":\"532 7\",\"pages\":\"\"},\"PeriodicalIF\":2.1000,\"publicationDate\":\"2024-07-23\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Comparative Neurology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/cne.25660\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"NEUROSCIENCES\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Comparative Neurology","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/cne.25660","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"NEUROSCIENCES","Score":null,"Total":0}
Clinicopathologic Dissociation: Robust Lafora Body Accumulation in Malin KO Mice Without Observable Changes in Home-Cage Behavior
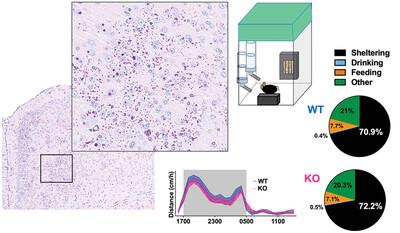
Lafora disease (LD) is a syndrome of progressive myoclonic epilepsy and cumulative neurocognitive deterioration caused by recessively inherited genetic lesions of EPM2A (laforin) or NHLRC1 (malin). Neuropsychiatric symptomatology in LD is thought to be directly downstream of neuronal and astrocytic polyglucosan aggregates, termed Lafora bodies (LBs), which faithfully accumulate in an age-dependent manner in all mouse models of LD. In this study, we applied home-cage monitoring to examine the extent of neurobehavioral deterioration in a model of malin-deficient LD as a means to identify robust preclinical endpoints that may guide the selection of novel genetic treatments. At 6 weeks, ∼6–7 months, and ∼12 months of age, malin-deficient mice (“KO”) and wild-type (WT) littermates underwent a standardized home-cage behavioral assessment designed to non-obtrusively appraise features of rest/arousal, consumptive behaviors, risk aversion, and voluntary wheel-running. At all timepoints, and over a range of metrics that we report transparently, WT and KO mice were essentially indistinguishable. In contrast, within WT mice compared across the same timepoints, we identified age-related nocturnal hypoactivity, diminished sucrose preference, and reduced wheel-running. Neuropathological examinations in subsets of the same mice revealed expected age-dependent LB accumulation, gliosis, and microglial activation in cortical and subcortical brain regions. At 12 months of age, despite the burden of neocortical LBs, we did not identify spontaneous seizures during an electroencephalographic (EEG) survey, and KO and WT mice exhibited similar spectral EEG features. However, in an in vitro assay of neocortical function, paroxysmal bursts of network activity (UP states) in KO slices were more prolonged at 3 and 6 months of age, but similar to WT at 12 months. KO mice displayed a distinct response to pentylenetetrazole, with a greater incidence of clonic seizures and a more pronounced postictal suppression of movement, feeding, and drinking behavior. Together, these results highlight the clinicopathologic dissociation in a mouse model of LD, where the accrual of LBs may latently modify cortical circuit function and seizure threshold without clinically meaningful changes in home-cage behavior. Our findings allude to a delay between LB accumulation and neurobehavioral decline in LD: one that may provide a window for treatment, and whose precise duration may be difficult to ascertain within the typical lifespan of a laboratory mouse.
期刊介绍:
Established in 1891, JCN is the oldest continually published basic neuroscience journal. Historically, as the name suggests, the journal focused on a comparison among species to uncover the intricacies of how the brain functions. In modern times, this research is called systems neuroscience where animal models are used to mimic core cognitive processes with the ultimate goal of understanding neural circuits and connections that give rise to behavioral patterns and different neural states.
Research published in JCN covers all species from invertebrates to humans, and the reports inform the readers about the function and organization of nervous systems in species with an emphasis on the way that species adaptations inform about the function or organization of the nervous systems, rather than on their evolution per se.
JCN publishes primary research articles and critical commentaries and review-type articles offering expert insight in to cutting edge research in the field of systems neuroscience; a complete list of contribution types is given in the Author Guidelines. For primary research contributions, only full-length investigative reports are desired; the journal does not accept short communications.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


