{"title":"多倍体中的变异调用,用于群体遗传学和数量遗传学","authors":"Alyssa R. Phillips","doi":"10.1002/aps3.11607","DOIUrl":null,"url":null,"abstract":"<p>Advancements in genome assembly and sequencing technology have made whole genome sequence (WGS) data and reference genomes accessible to study polyploid species. Compared to popular reduced-representation sequencing approaches, the genome-wide coverage and greater marker density provided by WGS data can greatly improve our understanding of polyploid species and polyploid biology. However, biological features that make polyploid species interesting also pose challenges in read mapping, variant identification, and genotype estimation. Accounting for characteristics in variant calling like allelic dosage uncertainty, homology between subgenomes, and variance in chromosome inheritance mode can reduce errors. Here, I discuss the challenges of variant calling in polyploid WGS data and discuss where potential solutions can be integrated into a standard variant calling pipeline.</p>","PeriodicalId":8022,"journal":{"name":"Applications in Plant Sciences","volume":"12 4","pages":""},"PeriodicalIF":2.4000,"publicationDate":"2024-07-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/aps3.11607","citationCount":"0","resultStr":"{\"title\":\"Variant calling in polyploids for population and quantitative genetics\",\"authors\":\"Alyssa R. Phillips\",\"doi\":\"10.1002/aps3.11607\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Advancements in genome assembly and sequencing technology have made whole genome sequence (WGS) data and reference genomes accessible to study polyploid species. Compared to popular reduced-representation sequencing approaches, the genome-wide coverage and greater marker density provided by WGS data can greatly improve our understanding of polyploid species and polyploid biology. However, biological features that make polyploid species interesting also pose challenges in read mapping, variant identification, and genotype estimation. Accounting for characteristics in variant calling like allelic dosage uncertainty, homology between subgenomes, and variance in chromosome inheritance mode can reduce errors. Here, I discuss the challenges of variant calling in polyploid WGS data and discuss where potential solutions can be integrated into a standard variant calling pipeline.</p>\",\"PeriodicalId\":8022,\"journal\":{\"name\":\"Applications in Plant Sciences\",\"volume\":\"12 4\",\"pages\":\"\"},\"PeriodicalIF\":2.4000,\"publicationDate\":\"2024-07-17\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/aps3.11607\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Applications in Plant Sciences\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://bsapubs.onlinelibrary.wiley.com/doi/10.1002/aps3.11607\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"PLANT SCIENCES\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Applications in Plant Sciences","FirstCategoryId":"99","ListUrlMain":"https://bsapubs.onlinelibrary.wiley.com/doi/10.1002/aps3.11607","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"PLANT SCIENCES","Score":null,"Total":0}
Variant calling in polyploids for population and quantitative genetics
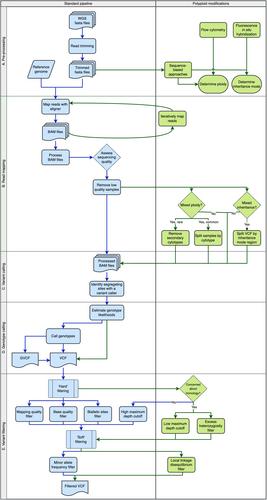
Advancements in genome assembly and sequencing technology have made whole genome sequence (WGS) data and reference genomes accessible to study polyploid species. Compared to popular reduced-representation sequencing approaches, the genome-wide coverage and greater marker density provided by WGS data can greatly improve our understanding of polyploid species and polyploid biology. However, biological features that make polyploid species interesting also pose challenges in read mapping, variant identification, and genotype estimation. Accounting for characteristics in variant calling like allelic dosage uncertainty, homology between subgenomes, and variance in chromosome inheritance mode can reduce errors. Here, I discuss the challenges of variant calling in polyploid WGS data and discuss where potential solutions can be integrated into a standard variant calling pipeline.
期刊介绍:
Applications in Plant Sciences (APPS) is a monthly, peer-reviewed, open access journal promoting the rapid dissemination of newly developed, innovative tools and protocols in all areas of the plant sciences, including genetics, structure, function, development, evolution, systematics, and ecology. Given the rapid progress today in technology and its application in the plant sciences, the goal of APPS is to foster communication within the plant science community to advance scientific research. APPS is a publication of the Botanical Society of America, originating in 2009 as the American Journal of Botany''s online-only section, AJB Primer Notes & Protocols in the Plant Sciences.
APPS publishes the following types of articles: (1) Protocol Notes describe new methods and technological advancements; (2) Genomic Resources Articles characterize the development and demonstrate the usefulness of newly developed genomic resources, including transcriptomes; (3) Software Notes detail new software applications; (4) Application Articles illustrate the application of a new protocol, method, or software application within the context of a larger study; (5) Review Articles evaluate available techniques, methods, or protocols; (6) Primer Notes report novel genetic markers with evidence of wide applicability.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


