{"title":"用于分子性质预测的原子注意力多体功能校正神经网络(MBNN-att)。","authors":"Zheng-Xin Yang, Xin-Tian Xie, Pei-Lin Kang, Zhen-Xiong Wang, Cheng Shang, Zhi-Pan Liu","doi":"10.1021/acs.jctc.4c00660","DOIUrl":null,"url":null,"abstract":"<p><p>Recent years have seen a surge of machine learning (ML) in chemistry for predicting chemical properties, but a low-cost, general-purpose, and high-performance model, desirable to be accessible on central processing unit (CPU) devices, remains not available. For this purpose, here we introduce an atomic attention mechanism into many-body function corrected neural network (MBNN), namely, MBNN-att ML model, to predict both the extensive and intensive properties of molecules and materials. The MBNN-att uses explicit function descriptors as the inputs for the atom-based feed-forward neural network (NN). The output of the NN is designed to be a vector to implement the multihead self-attention mechanism. This vector is split into two parts: the atomic attention weight part and the many-body-function part. The final property is obtained by summing the products of each atomic attention weight and the corresponding many-body function. We show that MBNN-att performs well on all QM9 properties, i.e., errors on all properties, below chemical accuracy, and, in particular, achieves the top performance for the energy-related extensive properties. By systematically comparing with other explicit-function-type descriptor ML models and the graph representation ML models, we demonstrate that the many-body-function framework and atomic attention mechanism are key ingredients for the high performance and the good transferability of MBNN-att in molecular property prediction.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":" ","pages":"6717-6727"},"PeriodicalIF":5.5000,"publicationDate":"2024-08-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Many-Body Function Corrected Neural Network with Atomic Attention (MBNN-att) for Molecular Property Prediction.\",\"authors\":\"Zheng-Xin Yang, Xin-Tian Xie, Pei-Lin Kang, Zhen-Xiong Wang, Cheng Shang, Zhi-Pan Liu\",\"doi\":\"10.1021/acs.jctc.4c00660\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Recent years have seen a surge of machine learning (ML) in chemistry for predicting chemical properties, but a low-cost, general-purpose, and high-performance model, desirable to be accessible on central processing unit (CPU) devices, remains not available. For this purpose, here we introduce an atomic attention mechanism into many-body function corrected neural network (MBNN), namely, MBNN-att ML model, to predict both the extensive and intensive properties of molecules and materials. The MBNN-att uses explicit function descriptors as the inputs for the atom-based feed-forward neural network (NN). The output of the NN is designed to be a vector to implement the multihead self-attention mechanism. This vector is split into two parts: the atomic attention weight part and the many-body-function part. The final property is obtained by summing the products of each atomic attention weight and the corresponding many-body function. We show that MBNN-att performs well on all QM9 properties, i.e., errors on all properties, below chemical accuracy, and, in particular, achieves the top performance for the energy-related extensive properties. By systematically comparing with other explicit-function-type descriptor ML models and the graph representation ML models, we demonstrate that the many-body-function framework and atomic attention mechanism are key ingredients for the high performance and the good transferability of MBNN-att in molecular property prediction.</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\" \",\"pages\":\"6717-6727\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2024-08-13\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jctc.4c00660\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/7/21 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c00660","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/7/21 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
摘要
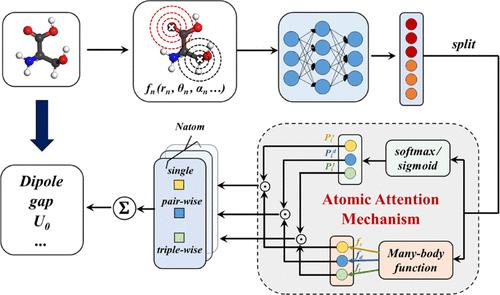
近年来,用于预测化学性质的机器学习(ML)技术在化学领域得到了蓬勃发展,但仍未出现一种可在中央处理器(CPU)设备上使用的低成本、通用且高性能的模型。为此,我们在多体函数校正神经网络(MBNN)中引入了原子注意机制,即 MBNN-att ML 模型,用于预测分子和材料的广义和狭义性质。MBNN-att 使用显式函数描述符作为基于原子的前馈神经网络(NN)的输入。神经网络的输出被设计为一个向量,以实现多头自注意机制。该向量分为两部分:原子注意力权重部分和多体功能部分。最终属性由每个原子注意力权重与相应的多体函数的乘积相加得出。我们的研究表明,MBNN-att 在所有 QM9 性质上都表现出色,即所有性质上的误差都低于化学精度,尤其是在与能量相关的广泛性质上表现出色。通过与其他显式函数型描述符 ML 模型和图表示 ML 模型进行系统比较,我们证明了多体函数框架和原子注意机制是 MBNN-att 在分子性质预测中实现高性能和良好可移植性的关键因素。
Many-Body Function Corrected Neural Network with Atomic Attention (MBNN-att) for Molecular Property Prediction.
Recent years have seen a surge of machine learning (ML) in chemistry for predicting chemical properties, but a low-cost, general-purpose, and high-performance model, desirable to be accessible on central processing unit (CPU) devices, remains not available. For this purpose, here we introduce an atomic attention mechanism into many-body function corrected neural network (MBNN), namely, MBNN-att ML model, to predict both the extensive and intensive properties of molecules and materials. The MBNN-att uses explicit function descriptors as the inputs for the atom-based feed-forward neural network (NN). The output of the NN is designed to be a vector to implement the multihead self-attention mechanism. This vector is split into two parts: the atomic attention weight part and the many-body-function part. The final property is obtained by summing the products of each atomic attention weight and the corresponding many-body function. We show that MBNN-att performs well on all QM9 properties, i.e., errors on all properties, below chemical accuracy, and, in particular, achieves the top performance for the energy-related extensive properties. By systematically comparing with other explicit-function-type descriptor ML models and the graph representation ML models, we demonstrate that the many-body-function framework and atomic attention mechanism are key ingredients for the high performance and the good transferability of MBNN-att in molecular property prediction.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


