Michelle L. Gaynor, Jacob B. Landis, Timothy K. O'Connor, Robert G. Laport, Jeff J. Doyle, Douglas E. Soltis, José Miguel Ponciano, Pamela S. Soltis
{"title":"nQuack:利用基于位点的杂合度从序列数据预测倍体水平的 R 软件包","authors":"Michelle L. Gaynor, Jacob B. Landis, Timothy K. O'Connor, Robert G. Laport, Jeff J. Doyle, Douglas E. Soltis, José Miguel Ponciano, Pamela S. Soltis","doi":"10.1002/aps3.11606","DOIUrl":null,"url":null,"abstract":"<div>\n \n \n <section>\n \n <h3> Premise</h3>\n \n <p>Traditional methods of ploidal-level estimation are tedious; using DNA sequence data for cytotype estimation is an ideal alternative. Multiple statistical approaches to leverage sequence data for ploidy inference based on site-based heterozygosity have been developed. However, these approaches may require high-coverage sequence data, use inappropriate probability distributions, or have additional statistical shortcomings that limit inference abilities. We introduce nQuack, an open-source R package that addresses the main shortcomings of current methods.</p>\n </section>\n \n <section>\n \n <h3> Methods and Results</h3>\n \n <p>nQuack performs model selection for improved ploidy predictions. Here, we implement expectation maximization algorithms with normal, beta, and beta-binomial distributions. Using extensive computer simulations that account for variability in sequencing depth, as well as real data sets, we demonstrate the utility and limitations of nQuack.</p>\n </section>\n \n <section>\n \n <h3> Conclusions</h3>\n \n <p>Inferring ploidy based on site-based heterozygosity alone is difficult. Even though nQuack is more accurate than similar methods, we suggest caution when relying on any site-based heterozygosity method to infer ploidy.</p>\n </section>\n </div>","PeriodicalId":8022,"journal":{"name":"Applications in Plant Sciences","volume":"12 4","pages":""},"PeriodicalIF":2.4000,"publicationDate":"2024-07-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/aps3.11606","citationCount":"0","resultStr":"{\"title\":\"nQuack: An R package for predicting ploidal level from sequence data using site-based heterozygosity\",\"authors\":\"Michelle L. Gaynor, Jacob B. Landis, Timothy K. O'Connor, Robert G. Laport, Jeff J. Doyle, Douglas E. Soltis, José Miguel Ponciano, Pamela S. Soltis\",\"doi\":\"10.1002/aps3.11606\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div>\\n \\n \\n <section>\\n \\n <h3> Premise</h3>\\n \\n <p>Traditional methods of ploidal-level estimation are tedious; using DNA sequence data for cytotype estimation is an ideal alternative. Multiple statistical approaches to leverage sequence data for ploidy inference based on site-based heterozygosity have been developed. However, these approaches may require high-coverage sequence data, use inappropriate probability distributions, or have additional statistical shortcomings that limit inference abilities. We introduce nQuack, an open-source R package that addresses the main shortcomings of current methods.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Methods and Results</h3>\\n \\n <p>nQuack performs model selection for improved ploidy predictions. Here, we implement expectation maximization algorithms with normal, beta, and beta-binomial distributions. Using extensive computer simulations that account for variability in sequencing depth, as well as real data sets, we demonstrate the utility and limitations of nQuack.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Conclusions</h3>\\n \\n <p>Inferring ploidy based on site-based heterozygosity alone is difficult. Even though nQuack is more accurate than similar methods, we suggest caution when relying on any site-based heterozygosity method to infer ploidy.</p>\\n </section>\\n </div>\",\"PeriodicalId\":8022,\"journal\":{\"name\":\"Applications in Plant Sciences\",\"volume\":\"12 4\",\"pages\":\"\"},\"PeriodicalIF\":2.4000,\"publicationDate\":\"2024-07-14\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/aps3.11606\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Applications in Plant Sciences\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://bsapubs.onlinelibrary.wiley.com/doi/10.1002/aps3.11606\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"PLANT SCIENCES\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Applications in Plant Sciences","FirstCategoryId":"99","ListUrlMain":"https://bsapubs.onlinelibrary.wiley.com/doi/10.1002/aps3.11606","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"PLANT SCIENCES","Score":null,"Total":0}
引用次数: 0
摘要
前提传统的倍性水平估算方法非常繁琐;利用 DNA 序列数据进行细胞型估算是一种理想的替代方法。目前已开发出多种统计方法,利用序列数据进行基于位点杂合度的倍性推断。然而,这些方法可能需要高覆盖率的序列数据、使用不恰当的概率分布,或存在其他限制推断能力的统计缺陷。我们介绍了一个开源 R 软件包 nQuack,它能解决当前方法的主要缺陷。方法与结果nQuack 能进行模型选择以改进倍性预测。在这里,我们使用正态分布、贝塔分布和贝塔二叉分布实现了期望最大化算法。结论仅根据基于位点的杂合度来推测倍性是很困难的。尽管 nQuack 比类似的方法更准确,但我们建议在依赖任何基于位点的杂合度方法来推断倍性时要谨慎。
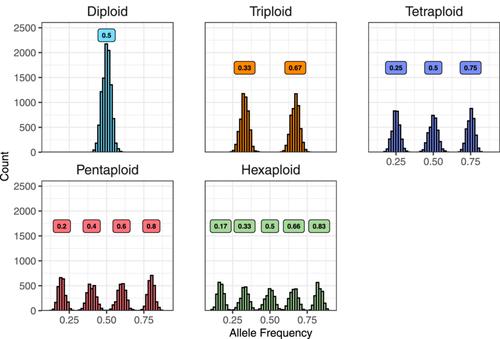
nQuack: An R package for predicting ploidal level from sequence data using site-based heterozygosity
Premise
Traditional methods of ploidal-level estimation are tedious; using DNA sequence data for cytotype estimation is an ideal alternative. Multiple statistical approaches to leverage sequence data for ploidy inference based on site-based heterozygosity have been developed. However, these approaches may require high-coverage sequence data, use inappropriate probability distributions, or have additional statistical shortcomings that limit inference abilities. We introduce nQuack, an open-source R package that addresses the main shortcomings of current methods.
Methods and Results
nQuack performs model selection for improved ploidy predictions. Here, we implement expectation maximization algorithms with normal, beta, and beta-binomial distributions. Using extensive computer simulations that account for variability in sequencing depth, as well as real data sets, we demonstrate the utility and limitations of nQuack.
Conclusions
Inferring ploidy based on site-based heterozygosity alone is difficult. Even though nQuack is more accurate than similar methods, we suggest caution when relying on any site-based heterozygosity method to infer ploidy.
期刊介绍:
Applications in Plant Sciences (APPS) is a monthly, peer-reviewed, open access journal promoting the rapid dissemination of newly developed, innovative tools and protocols in all areas of the plant sciences, including genetics, structure, function, development, evolution, systematics, and ecology. Given the rapid progress today in technology and its application in the plant sciences, the goal of APPS is to foster communication within the plant science community to advance scientific research. APPS is a publication of the Botanical Society of America, originating in 2009 as the American Journal of Botany''s online-only section, AJB Primer Notes & Protocols in the Plant Sciences.
APPS publishes the following types of articles: (1) Protocol Notes describe new methods and technological advancements; (2) Genomic Resources Articles characterize the development and demonstrate the usefulness of newly developed genomic resources, including transcriptomes; (3) Software Notes detail new software applications; (4) Application Articles illustrate the application of a new protocol, method, or software application within the context of a larger study; (5) Review Articles evaluate available techniques, methods, or protocols; (6) Primer Notes report novel genetic markers with evidence of wide applicability.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


