{"title":"IFT140 对无家族史多囊肾患者的重要性","authors":"Takuya Fujimaru , Takayasu Mori , Akinari Sekine , Motoko Chiga , Shintaro Mandai , Hiroaki Kikuchi , Yutaro Mori , Yu Hara , Tamami Fujiki , Fumiaki Ando , Koichiro Susa , Soichiro Iimori , Shotaro Naito , Ryoichi Hanazawa , Akihiro Hirakawa , Toshio Mochizuki , Tatsuya Suwabe , Yoshifumi Ubara , Shinichi Uchida , Eisei Sohara","doi":"10.1016/j.ekir.2024.06.021","DOIUrl":null,"url":null,"abstract":"<div><h3>Introduction</h3><p>Recently, the monoallelic loss-of-function IFT140 variant was identified as a causative gene for autosomal dominant polycystic kidney disease (ADPKD). In patients with polycystic kidneys who have a positive family history, >90% have pathogenic variants in <em>PKD1</em> or <em>PKD2</em>, whereas only 1% have <em>IFT140</em>. However, approximately 40% of patients with polycystic kidneys without a family history do not have any pathogenic variants in <em>PKD1</em> and <em>PKD2</em>.</p></div><div><h3>Methods</h3><p>We conducted a comprehensive genetic analysis of 157 adult patients with polycystic kidneys whose parents did not have evident polycystic kidneys. We sequenced up to 92 genes associated with inherited cystic kidney disease, including <em>IFT140</em>.</p></div><div><h3>Results</h3><p>Of the 157 patients, 7 (4.5%) presented with monoallelic loss-of-function variants in the <em>IFT140</em> gene, 51 (32.5%) with pathogenic variants in the <em>PKD1</em> or <em>PKD2</em> gene, and 7 (4.5%) with pathogenic variants in other genes related to inherited kidney cystic disease. The proportion of monoallelic loss-of-function <em>IFT140</em> variants in this cohort was higher than that in previously reported cohorts with polycystic kidneys who had a positive family history. None of the patients with monoallelic loss-of-function <em>IFT140</em> variants had polycystic liver disease (PLD). Furthermore, patients with <em>IFT140</em> pathogenic variants had a significantly smaller kidney volume and a remarkably higher estimated glomerular filtration rate (eGFR) than those with <em>PKD1</em> pathogenic variants (<em>P</em> = 0.01 and 0.03, respectively).</p></div><div><h3>Conclusion</h3><p>Because the phenotype of polycystic kidneys caused by the <em>IFT140</em> gene is mild, parental kidney disease may be overlooked. Therefore, patients without a positive family history are more likely to carry pathogenic variants in <em>IFT140</em>.</p></div>","PeriodicalId":17761,"journal":{"name":"Kidney International Reports","volume":"9 9","pages":"Pages 2685-2694"},"PeriodicalIF":5.7000,"publicationDate":"2024-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S2468024924017972/pdfft?md5=32c6c7c3e56f588bf020fd36e875b0a6&pid=1-s2.0-S2468024924017972-main.pdf","citationCount":"0","resultStr":"{\"title\":\"Importance of IFT140 in Patients with Polycystic Kidney Disease Without a Family History\",\"authors\":\"Takuya Fujimaru , Takayasu Mori , Akinari Sekine , Motoko Chiga , Shintaro Mandai , Hiroaki Kikuchi , Yutaro Mori , Yu Hara , Tamami Fujiki , Fumiaki Ando , Koichiro Susa , Soichiro Iimori , Shotaro Naito , Ryoichi Hanazawa , Akihiro Hirakawa , Toshio Mochizuki , Tatsuya Suwabe , Yoshifumi Ubara , Shinichi Uchida , Eisei Sohara\",\"doi\":\"10.1016/j.ekir.2024.06.021\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><h3>Introduction</h3><p>Recently, the monoallelic loss-of-function IFT140 variant was identified as a causative gene for autosomal dominant polycystic kidney disease (ADPKD). In patients with polycystic kidneys who have a positive family history, >90% have pathogenic variants in <em>PKD1</em> or <em>PKD2</em>, whereas only 1% have <em>IFT140</em>. However, approximately 40% of patients with polycystic kidneys without a family history do not have any pathogenic variants in <em>PKD1</em> and <em>PKD2</em>.</p></div><div><h3>Methods</h3><p>We conducted a comprehensive genetic analysis of 157 adult patients with polycystic kidneys whose parents did not have evident polycystic kidneys. We sequenced up to 92 genes associated with inherited cystic kidney disease, including <em>IFT140</em>.</p></div><div><h3>Results</h3><p>Of the 157 patients, 7 (4.5%) presented with monoallelic loss-of-function variants in the <em>IFT140</em> gene, 51 (32.5%) with pathogenic variants in the <em>PKD1</em> or <em>PKD2</em> gene, and 7 (4.5%) with pathogenic variants in other genes related to inherited kidney cystic disease. The proportion of monoallelic loss-of-function <em>IFT140</em> variants in this cohort was higher than that in previously reported cohorts with polycystic kidneys who had a positive family history. None of the patients with monoallelic loss-of-function <em>IFT140</em> variants had polycystic liver disease (PLD). Furthermore, patients with <em>IFT140</em> pathogenic variants had a significantly smaller kidney volume and a remarkably higher estimated glomerular filtration rate (eGFR) than those with <em>PKD1</em> pathogenic variants (<em>P</em> = 0.01 and 0.03, respectively).</p></div><div><h3>Conclusion</h3><p>Because the phenotype of polycystic kidneys caused by the <em>IFT140</em> gene is mild, parental kidney disease may be overlooked. Therefore, patients without a positive family history are more likely to carry pathogenic variants in <em>IFT140</em>.</p></div>\",\"PeriodicalId\":17761,\"journal\":{\"name\":\"Kidney International Reports\",\"volume\":\"9 9\",\"pages\":\"Pages 2685-2694\"},\"PeriodicalIF\":5.7000,\"publicationDate\":\"2024-09-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.sciencedirect.com/science/article/pii/S2468024924017972/pdfft?md5=32c6c7c3e56f588bf020fd36e875b0a6&pid=1-s2.0-S2468024924017972-main.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Kidney International Reports\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S2468024924017972\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"UROLOGY & NEPHROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Kidney International Reports","FirstCategoryId":"3","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2468024924017972","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"UROLOGY & NEPHROLOGY","Score":null,"Total":0}
Importance of IFT140 in Patients with Polycystic Kidney Disease Without a Family History
Introduction
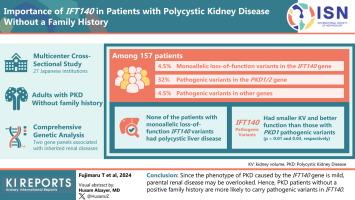
Recently, the monoallelic loss-of-function IFT140 variant was identified as a causative gene for autosomal dominant polycystic kidney disease (ADPKD). In patients with polycystic kidneys who have a positive family history, >90% have pathogenic variants in PKD1 or PKD2, whereas only 1% have IFT140. However, approximately 40% of patients with polycystic kidneys without a family history do not have any pathogenic variants in PKD1 and PKD2.
Methods
We conducted a comprehensive genetic analysis of 157 adult patients with polycystic kidneys whose parents did not have evident polycystic kidneys. We sequenced up to 92 genes associated with inherited cystic kidney disease, including IFT140.
Results
Of the 157 patients, 7 (4.5%) presented with monoallelic loss-of-function variants in the IFT140 gene, 51 (32.5%) with pathogenic variants in the PKD1 or PKD2 gene, and 7 (4.5%) with pathogenic variants in other genes related to inherited kidney cystic disease. The proportion of monoallelic loss-of-function IFT140 variants in this cohort was higher than that in previously reported cohorts with polycystic kidneys who had a positive family history. None of the patients with monoallelic loss-of-function IFT140 variants had polycystic liver disease (PLD). Furthermore, patients with IFT140 pathogenic variants had a significantly smaller kidney volume and a remarkably higher estimated glomerular filtration rate (eGFR) than those with PKD1 pathogenic variants (P = 0.01 and 0.03, respectively).
Conclusion
Because the phenotype of polycystic kidneys caused by the IFT140 gene is mild, parental kidney disease may be overlooked. Therefore, patients without a positive family history are more likely to carry pathogenic variants in IFT140.
期刊介绍:
Kidney International Reports, an official journal of the International Society of Nephrology, is a peer-reviewed, open access journal devoted to the publication of leading research and developments related to kidney disease. With the primary aim of contributing to improved care of patients with kidney disease, the journal will publish original clinical and select translational articles and educational content related to the pathogenesis, evaluation and management of acute and chronic kidney disease, end stage renal disease (including transplantation), acid-base, fluid and electrolyte disturbances and hypertension. Of particular interest are submissions related to clinical trials, epidemiology, systematic reviews (including meta-analyses) and outcomes research. The journal will also provide a platform for wider dissemination of national and regional guidelines as well as consensus meeting reports.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


