{"title":"印度喜马拉雅地区一家偏远山区医院的一例罕见零星克雅二氏症病例。","authors":"Nitu Sharma, Jitender Kumar Sharma, Ashima Chander, Khushdeep Shergill, Meghna Yadav","doi":"10.4322/acr.2024.502","DOIUrl":null,"url":null,"abstract":"<p><p>Sporadic Creutzfeldt-Jakob disease (CJD) is a rare neurodegenerative spongiform encephalopathy that causes neuronal derangement secondary to prion protein. Its initial diagnosis is often complex and challenging due to non-specific clinical presentation, lack of awareness, and low clinical suspicion. This disease is invariably fatal, and most patients die within 12 months of presentation. Definite diagnosis of prion disease requires neuropathological analysis, usually done at autopsy. Here, we present the autopsy findings of a 57-year-old male patient, illustrating the complexity of diagnosing this disease early in the clinical course and the need for a broad differential diagnosis at the onset.</p>","PeriodicalId":53117,"journal":{"name":"Autopsy and Case Reports","volume":"14 ","pages":"e2024502"},"PeriodicalIF":0.0000,"publicationDate":"2024-06-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11253907/pdf/","citationCount":"0","resultStr":"{\"title\":\"A rare case of Sporadic Creutzfeldt-Jakob disease at a remote mountain hospital in the Indian Himalayan Region.\",\"authors\":\"Nitu Sharma, Jitender Kumar Sharma, Ashima Chander, Khushdeep Shergill, Meghna Yadav\",\"doi\":\"10.4322/acr.2024.502\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Sporadic Creutzfeldt-Jakob disease (CJD) is a rare neurodegenerative spongiform encephalopathy that causes neuronal derangement secondary to prion protein. Its initial diagnosis is often complex and challenging due to non-specific clinical presentation, lack of awareness, and low clinical suspicion. This disease is invariably fatal, and most patients die within 12 months of presentation. Definite diagnosis of prion disease requires neuropathological analysis, usually done at autopsy. Here, we present the autopsy findings of a 57-year-old male patient, illustrating the complexity of diagnosing this disease early in the clinical course and the need for a broad differential diagnosis at the onset.</p>\",\"PeriodicalId\":53117,\"journal\":{\"name\":\"Autopsy and Case Reports\",\"volume\":\"14 \",\"pages\":\"e2024502\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2024-06-21\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11253907/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Autopsy and Case Reports\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.4322/acr.2024.502\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q4\",\"JCRName\":\"Medicine\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Autopsy and Case Reports","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.4322/acr.2024.502","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"Medicine","Score":null,"Total":0}
A rare case of Sporadic Creutzfeldt-Jakob disease at a remote mountain hospital in the Indian Himalayan Region.
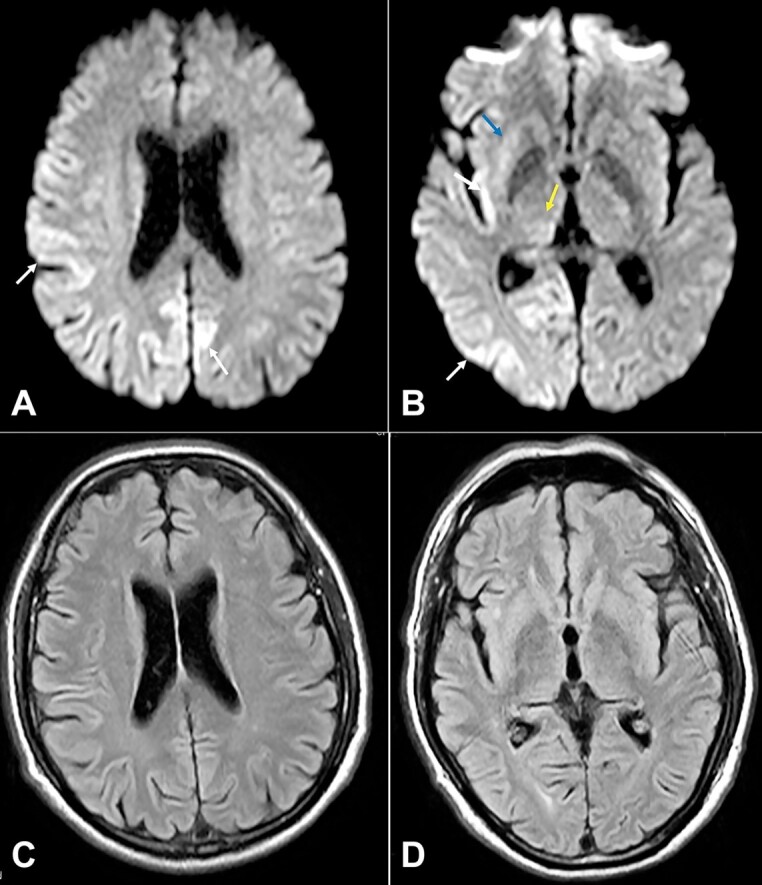
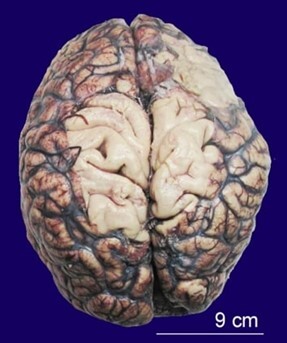
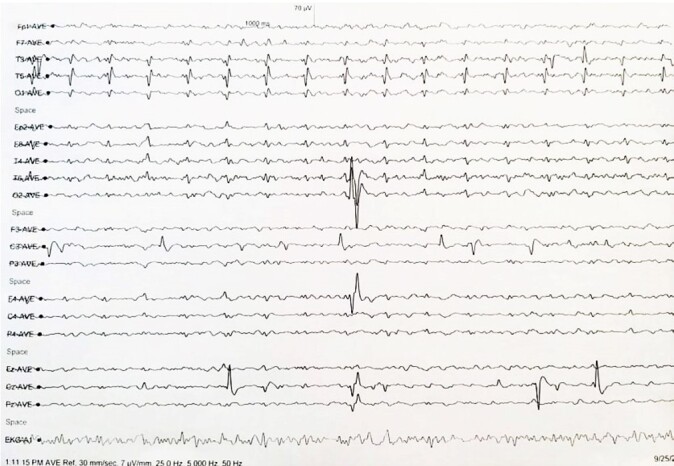
Sporadic Creutzfeldt-Jakob disease (CJD) is a rare neurodegenerative spongiform encephalopathy that causes neuronal derangement secondary to prion protein. Its initial diagnosis is often complex and challenging due to non-specific clinical presentation, lack of awareness, and low clinical suspicion. This disease is invariably fatal, and most patients die within 12 months of presentation. Definite diagnosis of prion disease requires neuropathological analysis, usually done at autopsy. Here, we present the autopsy findings of a 57-year-old male patient, illustrating the complexity of diagnosing this disease early in the clinical course and the need for a broad differential diagnosis at the onset.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


