Shuai Wang, Gengmiao Xiao, Minyi Tang, Xinyun Bi, Chaofeng Xing, Aolu Liu, Allan Z Zhao, Fanghong Li
{"title":"FKBP38 基因缺失会通过 MCP-1/p38 通路促进免疫反应,从而加剧 ConA 诱导的肝炎。","authors":"Shuai Wang, Gengmiao Xiao, Minyi Tang, Xinyun Bi, Chaofeng Xing, Aolu Liu, Allan Z Zhao, Fanghong Li","doi":"10.1016/j.intimp.2024.112659","DOIUrl":null,"url":null,"abstract":"<p><p>Autoimmune hepatitis (AIH) is a chronic liver disease characterized by immune dysregulation and hepatocyte damage. FKBP38, a member of the immunophilin family, has been implicated in immune regulation and the modulation of intracellular signaling pathways; however, its role in AIH pathogenesis remains poorly understood. In this study, we aimed to investigate the effects of hepatic FKBP38 deletion on AIH using a hepatic FKBP38 knockout (LKO) mouse model created via cre-loxP technology. We compared the survival rates, incidence, and severity of AIH in LKO mice with those in control mice. Our findings revealed that hepatic FKBP38 deletion resulted in an unfavorable prognosis in LKO mice with AIH. Specifically, LKO mice exhibited heightened liver inflammation and extensive hepatocyte damage compared to control mice, with a significant decrease in anti-apoptotic proteins and a marked increase in pro-apoptotic proteins. Additionally, transcriptional and translational levels of pro-inflammatory cytokines and chemokines were significantly increased in LKO mice compared to control mice. Immunoblot analysis showed that MCP-1 expression was significantly elevated in LKO mice. Furthermore, the phosphorylation of p38 was increased in LKO mice with AIH, indicating that FKBP38 deletion promotes liver injury in AIH by upregulating p38 phosphorylation and increasing MCP-1 expression. Immune cell profiling demonstrated elevated populations of T, NK, and B cells, suggesting a dysregulated immune response in LKO mice with AIH. Overall, our findings suggest that FKBP38 disruption exacerbates AIH severity by augmenting the immune response by activating the MCP-1/p38 signaling pathway.</p>","PeriodicalId":13859,"journal":{"name":"International immunopharmacology","volume":null,"pages":null},"PeriodicalIF":4.8000,"publicationDate":"2024-09-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"FKBP38 deletion exacerbates ConA-induced hepatitis by promoting the immune response through the MCP-1/p38 pathway.\",\"authors\":\"Shuai Wang, Gengmiao Xiao, Minyi Tang, Xinyun Bi, Chaofeng Xing, Aolu Liu, Allan Z Zhao, Fanghong Li\",\"doi\":\"10.1016/j.intimp.2024.112659\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Autoimmune hepatitis (AIH) is a chronic liver disease characterized by immune dysregulation and hepatocyte damage. FKBP38, a member of the immunophilin family, has been implicated in immune regulation and the modulation of intracellular signaling pathways; however, its role in AIH pathogenesis remains poorly understood. In this study, we aimed to investigate the effects of hepatic FKBP38 deletion on AIH using a hepatic FKBP38 knockout (LKO) mouse model created via cre-loxP technology. We compared the survival rates, incidence, and severity of AIH in LKO mice with those in control mice. Our findings revealed that hepatic FKBP38 deletion resulted in an unfavorable prognosis in LKO mice with AIH. Specifically, LKO mice exhibited heightened liver inflammation and extensive hepatocyte damage compared to control mice, with a significant decrease in anti-apoptotic proteins and a marked increase in pro-apoptotic proteins. Additionally, transcriptional and translational levels of pro-inflammatory cytokines and chemokines were significantly increased in LKO mice compared to control mice. Immunoblot analysis showed that MCP-1 expression was significantly elevated in LKO mice. Furthermore, the phosphorylation of p38 was increased in LKO mice with AIH, indicating that FKBP38 deletion promotes liver injury in AIH by upregulating p38 phosphorylation and increasing MCP-1 expression. Immune cell profiling demonstrated elevated populations of T, NK, and B cells, suggesting a dysregulated immune response in LKO mice with AIH. Overall, our findings suggest that FKBP38 disruption exacerbates AIH severity by augmenting the immune response by activating the MCP-1/p38 signaling pathway.</p>\",\"PeriodicalId\":13859,\"journal\":{\"name\":\"International immunopharmacology\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":4.8000,\"publicationDate\":\"2024-09-10\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"International immunopharmacology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1016/j.intimp.2024.112659\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/7/11 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"IMMUNOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"International immunopharmacology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1016/j.intimp.2024.112659","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/7/11 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"IMMUNOLOGY","Score":null,"Total":0}
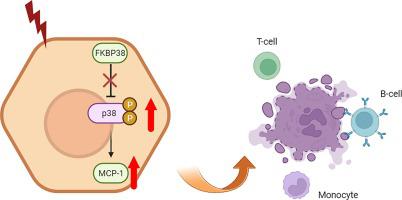
FKBP38 deletion exacerbates ConA-induced hepatitis by promoting the immune response through the MCP-1/p38 pathway.
Autoimmune hepatitis (AIH) is a chronic liver disease characterized by immune dysregulation and hepatocyte damage. FKBP38, a member of the immunophilin family, has been implicated in immune regulation and the modulation of intracellular signaling pathways; however, its role in AIH pathogenesis remains poorly understood. In this study, we aimed to investigate the effects of hepatic FKBP38 deletion on AIH using a hepatic FKBP38 knockout (LKO) mouse model created via cre-loxP technology. We compared the survival rates, incidence, and severity of AIH in LKO mice with those in control mice. Our findings revealed that hepatic FKBP38 deletion resulted in an unfavorable prognosis in LKO mice with AIH. Specifically, LKO mice exhibited heightened liver inflammation and extensive hepatocyte damage compared to control mice, with a significant decrease in anti-apoptotic proteins and a marked increase in pro-apoptotic proteins. Additionally, transcriptional and translational levels of pro-inflammatory cytokines and chemokines were significantly increased in LKO mice compared to control mice. Immunoblot analysis showed that MCP-1 expression was significantly elevated in LKO mice. Furthermore, the phosphorylation of p38 was increased in LKO mice with AIH, indicating that FKBP38 deletion promotes liver injury in AIH by upregulating p38 phosphorylation and increasing MCP-1 expression. Immune cell profiling demonstrated elevated populations of T, NK, and B cells, suggesting a dysregulated immune response in LKO mice with AIH. Overall, our findings suggest that FKBP38 disruption exacerbates AIH severity by augmenting the immune response by activating the MCP-1/p38 signaling pathway.
期刊介绍:
International Immunopharmacology is the primary vehicle for the publication of original research papers pertinent to the overlapping areas of immunology, pharmacology, cytokine biology, immunotherapy, immunopathology and immunotoxicology. Review articles that encompass these subjects are also welcome.
The subject material appropriate for submission includes:
• Clinical studies employing immunotherapy of any type including the use of: bacterial and chemical agents; thymic hormones, interferon, lymphokines, etc., in transplantation and diseases such as cancer, immunodeficiency, chronic infection and allergic, inflammatory or autoimmune disorders.
• Studies on the mechanisms of action of these agents for specific parameters of immune competence as well as the overall clinical state.
• Pre-clinical animal studies and in vitro studies on mechanisms of action with immunopotentiators, immunomodulators, immunoadjuvants and other pharmacological agents active on cells participating in immune or allergic responses.
• Pharmacological compounds, microbial products and toxicological agents that affect the lymphoid system, and their mechanisms of action.
• Agents that activate genes or modify transcription and translation within the immune response.
• Substances activated, generated, or released through immunologic or related pathways that are pharmacologically active.
• Production, function and regulation of cytokines and their receptors.
• Classical pharmacological studies on the effects of chemokines and bioactive factors released during immunological reactions.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


