Raphael O. Betschart, Cristian Riccio, Domingo Aguilera-Garcia, Stefan Blankenberg, Linlin Guo, Holger Moch, Dagmar Seidl, Hugo Solleder, Felix Thalén, Alexandre Thiéry, Raphael Twerenbold, Tanja Zeller, Martin Zoche, Andreas Ziegler
{"title":"全基因组测序研究的生物统计方面:预处理和质量控制","authors":"Raphael O. Betschart, Cristian Riccio, Domingo Aguilera-Garcia, Stefan Blankenberg, Linlin Guo, Holger Moch, Dagmar Seidl, Hugo Solleder, Felix Thalén, Alexandre Thiéry, Raphael Twerenbold, Tanja Zeller, Martin Zoche, Andreas Ziegler","doi":"10.1002/bimj.202300278","DOIUrl":null,"url":null,"abstract":"<p>Rapid advances in high-throughput DNA sequencing technologies have enabled large-scale whole genome sequencing (WGS) studies. Before performing association analysis between phenotypes and genotypes, preprocessing and quality control (QC) of the raw sequence data need to be performed. Because many biostatisticians have not been working with WGS data so far, we first sketch Illumina's short-read sequencing technology. Second, we explain the general preprocessing pipeline for WGS studies. Third, we provide an overview of important QC metrics, which are applied to WGS data: on the raw data, after mapping and alignment, after variant calling, and after multisample variant calling. Fourth, we illustrate the QC with the data from the GENEtic SequencIng Study Hamburg–Davos (GENESIS-HD), a study involving more than 9000 human whole genomes. All samples were sequenced on an Illumina NovaSeq 6000 with an average coverage of 35× using a PCR-free protocol. For QC, one genome in a bottle (GIAB) trio was sequenced in four replicates, and one GIAB sample was successfully sequenced 70 times in different runs. Fifth, we provide empirical data on the compression of raw data using the DRAGEN original read archive (ORA). The most important quality metrics in the application were genetic similarity, sample cross-contamination, deviations from the expected Het/Hom ratio, relatedness, and coverage. The compression ratio of the raw files using DRAGEN ORA was 5.6:1, and compression time was linear by genome coverage. In summary, the preprocessing, joint calling, and QC of large WGS studies are feasible within a reasonable time, and efficient QC procedures are readily available.</p>","PeriodicalId":55360,"journal":{"name":"Biometrical Journal","volume":"66 5","pages":""},"PeriodicalIF":1.3000,"publicationDate":"2024-07-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/bimj.202300278","citationCount":"0","resultStr":"{\"title\":\"Biostatistical Aspects of Whole Genome Sequencing Studies: Preprocessing and Quality Control\",\"authors\":\"Raphael O. Betschart, Cristian Riccio, Domingo Aguilera-Garcia, Stefan Blankenberg, Linlin Guo, Holger Moch, Dagmar Seidl, Hugo Solleder, Felix Thalén, Alexandre Thiéry, Raphael Twerenbold, Tanja Zeller, Martin Zoche, Andreas Ziegler\",\"doi\":\"10.1002/bimj.202300278\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Rapid advances in high-throughput DNA sequencing technologies have enabled large-scale whole genome sequencing (WGS) studies. Before performing association analysis between phenotypes and genotypes, preprocessing and quality control (QC) of the raw sequence data need to be performed. Because many biostatisticians have not been working with WGS data so far, we first sketch Illumina's short-read sequencing technology. Second, we explain the general preprocessing pipeline for WGS studies. Third, we provide an overview of important QC metrics, which are applied to WGS data: on the raw data, after mapping and alignment, after variant calling, and after multisample variant calling. Fourth, we illustrate the QC with the data from the GENEtic SequencIng Study Hamburg–Davos (GENESIS-HD), a study involving more than 9000 human whole genomes. All samples were sequenced on an Illumina NovaSeq 6000 with an average coverage of 35× using a PCR-free protocol. For QC, one genome in a bottle (GIAB) trio was sequenced in four replicates, and one GIAB sample was successfully sequenced 70 times in different runs. Fifth, we provide empirical data on the compression of raw data using the DRAGEN original read archive (ORA). The most important quality metrics in the application were genetic similarity, sample cross-contamination, deviations from the expected Het/Hom ratio, relatedness, and coverage. The compression ratio of the raw files using DRAGEN ORA was 5.6:1, and compression time was linear by genome coverage. In summary, the preprocessing, joint calling, and QC of large WGS studies are feasible within a reasonable time, and efficient QC procedures are readily available.</p>\",\"PeriodicalId\":55360,\"journal\":{\"name\":\"Biometrical Journal\",\"volume\":\"66 5\",\"pages\":\"\"},\"PeriodicalIF\":1.3000,\"publicationDate\":\"2024-07-11\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/bimj.202300278\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Biometrical Journal\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/bimj.202300278\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"MATHEMATICAL & COMPUTATIONAL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biometrical Journal","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/bimj.202300278","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
Biostatistical Aspects of Whole Genome Sequencing Studies: Preprocessing and Quality Control
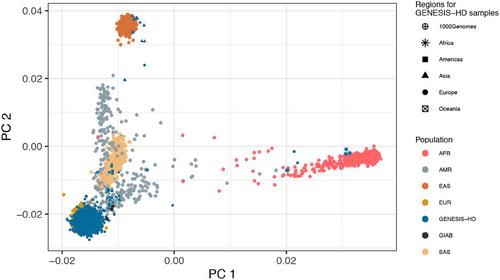
Rapid advances in high-throughput DNA sequencing technologies have enabled large-scale whole genome sequencing (WGS) studies. Before performing association analysis between phenotypes and genotypes, preprocessing and quality control (QC) of the raw sequence data need to be performed. Because many biostatisticians have not been working with WGS data so far, we first sketch Illumina's short-read sequencing technology. Second, we explain the general preprocessing pipeline for WGS studies. Third, we provide an overview of important QC metrics, which are applied to WGS data: on the raw data, after mapping and alignment, after variant calling, and after multisample variant calling. Fourth, we illustrate the QC with the data from the GENEtic SequencIng Study Hamburg–Davos (GENESIS-HD), a study involving more than 9000 human whole genomes. All samples were sequenced on an Illumina NovaSeq 6000 with an average coverage of 35× using a PCR-free protocol. For QC, one genome in a bottle (GIAB) trio was sequenced in four replicates, and one GIAB sample was successfully sequenced 70 times in different runs. Fifth, we provide empirical data on the compression of raw data using the DRAGEN original read archive (ORA). The most important quality metrics in the application were genetic similarity, sample cross-contamination, deviations from the expected Het/Hom ratio, relatedness, and coverage. The compression ratio of the raw files using DRAGEN ORA was 5.6:1, and compression time was linear by genome coverage. In summary, the preprocessing, joint calling, and QC of large WGS studies are feasible within a reasonable time, and efficient QC procedures are readily available.
期刊介绍:
Biometrical Journal publishes papers on statistical methods and their applications in life sciences including medicine, environmental sciences and agriculture. Methodological developments should be motivated by an interesting and relevant problem from these areas. Ideally the manuscript should include a description of the problem and a section detailing the application of the new methodology to the problem. Case studies, review articles and letters to the editors are also welcome. Papers containing only extensive mathematical theory are not suitable for publication in Biometrical Journal.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


