突尼斯 COG5-CDG 综合征患者 COG5 中一个错义变体的特征以及非同义变体对 COG5 蛋白质影响的见解。
IF 2.6
3区 生物学
Q2 GENETICS & HEREDITY
引用次数: 0
摘要
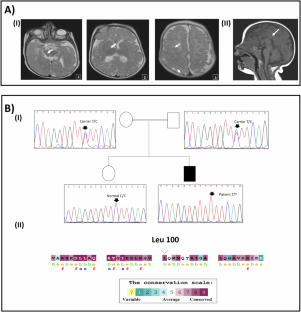
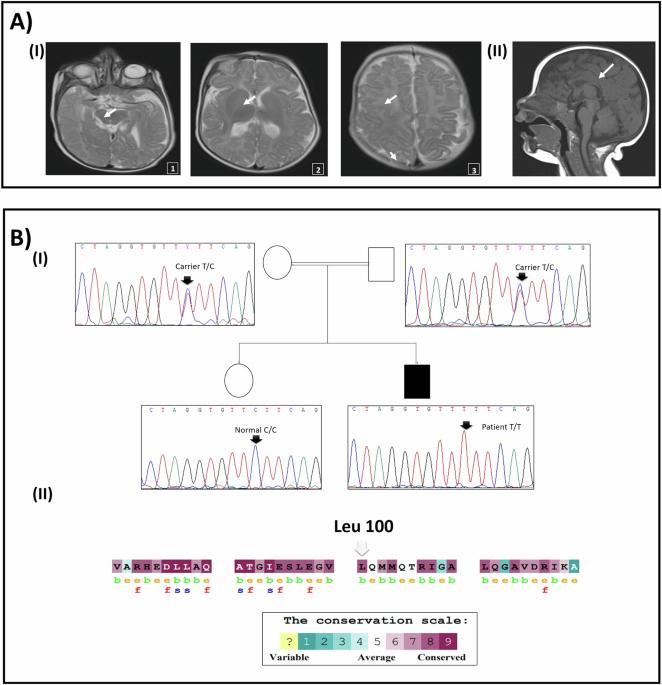
多系统受累(包括明显的神经系统损伤)患者的临床诊断具有挑战性。高通量测序方法对于提供准确诊断至关重要。在本研究中,我们报告了一名表现为肌张力低下、全身发育迟缓并伴有视觉和皮肤异常的突尼斯患者。我们进行了外显子组测序,随后进行了分离分析和其他调查。研究人员对 COG5 保守位置上的非同义变异(nsSNPs)进行了硅分析。结果发现,COG5 中有一个同源错义变异 c.298 C > T(p.Leu100Phe),遗传自父母双方。该变异不仅改变了 COG5-COG7 的相互作用,还改变了蛋白质的溶解度和稳定性。在体外 COG5 协同免疫沉淀中,使用患者衍生细胞证实了这种相互作用的中断,在这种情况下,与结合伙伴 COG7 的相互作用被削弱。因此,我们确定了 COG5-CDG 的诊断。从临床上看,该患者与已描述过的病例具有共同特征,但恙虫病是一种新的表现。相反,CADD 评分显示了 19 个可能致病的 nsSNPs(最小等位基因频率 MAF 30),其中 11 个对 COG5 的溶解度和/或稳定性有显著影响。这些特性似乎被七个错义 COG5-CDG 变异中的六个所破坏。总之,我们的研究扩大了 COG5-CDG 疾病的遗传和表型谱,并强调了新一代测序作为精确诊断的强大工具的实用性。我们的研究结果揭示了COG5错义变异致病的可能分子机制,即COG5稳定性和溶解性的改变。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Characterization of a missense variant in COG5 in a Tunisian patient with COG5-CDG syndrome and insights into the effect of non-synonymous variants on COG5 protein
The clinical diagnosis of patients with multisystem involvement including a pronounced neurologic damage is challenging. High-throughput sequencing methods remains crucial to provide an accurate diagnosis. In this study, we reported a Tunisian patient manifesting hypotonia and global developmental delay with visual and skin abnormalities. Exome sequencing was conducted followed by segregation analysis and, subsequently additional investigations. In silico analysis of non-synonymous variants (nsSNPs) described in COG5 in conserved positions was made. Results revealed a homozygous missense variant c.298 C > T (p.Leu100Phe) in the COG5 inherited from both parents. This variant altered both protein solubility and stability, in addition to a putative disruption of the COG5-COG7 interaction. This disruption has been confirmed using patient-derived cells in vitro in a COG5 co-immuno-precipitation, where interaction with binding partner COG7 was abrogated. Hence, we established the COG5-CDG diagnosis. Clinically, the patient shared common features with the already described cases with the report of the ichtyosis as a new manifestation. Conversely, the CADD scoring revealed 19 putatively pathogenic nsSNPs (Minor Allele Frequency MAF < 0.001, CADD > 30), 11 of which had a significant impact on the solubility and/or stability of COG5. These properties seem to be disrupted by six of the seven missense COG5-CDG variants. In conclusion, our study expands the genetic and phenotypic spectrum of COG5-CDG disease and highlight the utility of the next generation sequencing as a powerful tool in accurate diagnosis. Our results shed light on a likely molecular mechanism underlying the pathogenic effect of missense COG5 variants, which is the alteration of COG5 stability and solubility.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Journal of Human Genetics
生物-遗传学
CiteScore
7.20
自引率
0.00%
发文量
101
审稿时长
4-8 weeks
期刊介绍:
The Journal of Human Genetics is an international journal publishing articles on human genetics, including medical genetics and human genome analysis. It covers all aspects of human genetics, including molecular genetics, clinical genetics, behavioral genetics, immunogenetics, pharmacogenomics, population genetics, functional genomics, epigenetics, genetic counseling and gene therapy.
Articles on the following areas are especially welcome: genetic factors of monogenic and complex disorders, genome-wide association studies, genetic epidemiology, cancer genetics, personal genomics, genotype-phenotype relationships and genome diversity.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


