Yan Zhang, Shota Fukuma, Rui Shang, Eiichi Nakamura
{"title":"铁催化 C(sp²)-H活化,在扩展的π-共轭体系上与炔烃进行偶氮annulation反应","authors":"Yan Zhang, Shota Fukuma, Rui Shang, Eiichi Nakamura","doi":"10.1038/s44160-024-00605-6","DOIUrl":null,"url":null,"abstract":"Sustainable C–H activation methods for expanding large π-systems, crucial for electronic materials, continue to pose a challenge due to the metal–π interaction involving bonding and back-bonding. Here we present an iron-catalysed method for aza-annulation π-extension reactions of conjugated carbonyl compounds via oxime ether, employing a sterically bulky trisphosphine ligand to mitigate Fe–π interaction. Inexpensive isobutyl aluminium(III) catecholate serves as a base. These reactions convert readily available substrates, such as quinacridone, pentacenedione and pentacenetetraone, into conjugated skeletons, yielding several narrow-band-emissive molecules, including an actively pursued deep-blue emitter with peak emission at 450 nm and a narrow emission band of 13 nm. The tetrafold C–H activation of pentacenetetraone, with each step achieving an average yield of 95%, underscores the efficacy of iron catalysis in selectively activating C–H bonds on π-extended systems, offering promise for expanding organic electronic materials. Metal–π interactions typically hinder metal-catalysed C–H functionalization reactions involving large π-conjugated systems. Now, an iron-catalysed aza-annulation method, employing a bulky trisphosphine ligand and an aluminium base, proves efficient for large π-extended substrates, holding promise for electronic materials discovery.","PeriodicalId":74251,"journal":{"name":"Nature synthesis","volume":"3 11","pages":"1349-1359"},"PeriodicalIF":0.0000,"publicationDate":"2024-07-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Iron-catalysed C(sp²)–H activation for aza-annulation with alkynes on extended π-conjugated systems\",\"authors\":\"Yan Zhang, Shota Fukuma, Rui Shang, Eiichi Nakamura\",\"doi\":\"10.1038/s44160-024-00605-6\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Sustainable C–H activation methods for expanding large π-systems, crucial for electronic materials, continue to pose a challenge due to the metal–π interaction involving bonding and back-bonding. Here we present an iron-catalysed method for aza-annulation π-extension reactions of conjugated carbonyl compounds via oxime ether, employing a sterically bulky trisphosphine ligand to mitigate Fe–π interaction. Inexpensive isobutyl aluminium(III) catecholate serves as a base. These reactions convert readily available substrates, such as quinacridone, pentacenedione and pentacenetetraone, into conjugated skeletons, yielding several narrow-band-emissive molecules, including an actively pursued deep-blue emitter with peak emission at 450 nm and a narrow emission band of 13 nm. The tetrafold C–H activation of pentacenetetraone, with each step achieving an average yield of 95%, underscores the efficacy of iron catalysis in selectively activating C–H bonds on π-extended systems, offering promise for expanding organic electronic materials. Metal–π interactions typically hinder metal-catalysed C–H functionalization reactions involving large π-conjugated systems. Now, an iron-catalysed aza-annulation method, employing a bulky trisphosphine ligand and an aluminium base, proves efficient for large π-extended substrates, holding promise for electronic materials discovery.\",\"PeriodicalId\":74251,\"journal\":{\"name\":\"Nature synthesis\",\"volume\":\"3 11\",\"pages\":\"1349-1359\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2024-07-09\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Nature synthesis\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://www.nature.com/articles/s44160-024-00605-6\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"0\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature synthesis","FirstCategoryId":"1085","ListUrlMain":"https://www.nature.com/articles/s44160-024-00605-6","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"0","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Iron-catalysed C(sp²)–H activation for aza-annulation with alkynes on extended π-conjugated systems
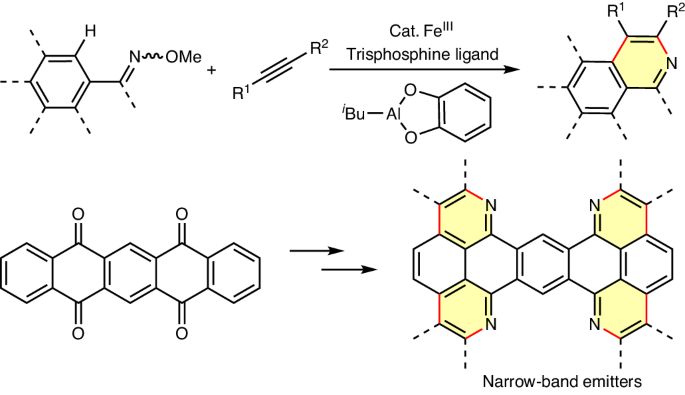
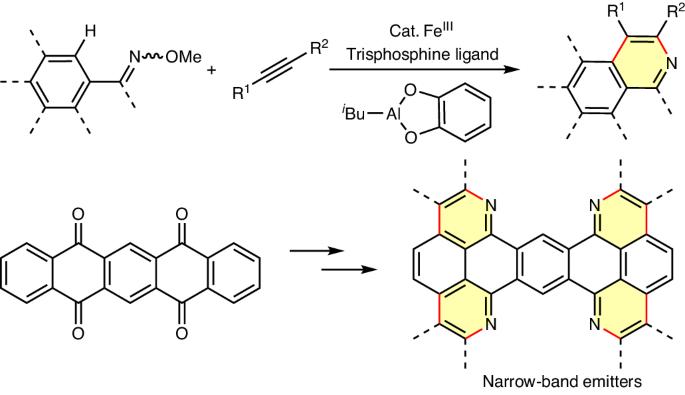
Sustainable C–H activation methods for expanding large π-systems, crucial for electronic materials, continue to pose a challenge due to the metal–π interaction involving bonding and back-bonding. Here we present an iron-catalysed method for aza-annulation π-extension reactions of conjugated carbonyl compounds via oxime ether, employing a sterically bulky trisphosphine ligand to mitigate Fe–π interaction. Inexpensive isobutyl aluminium(III) catecholate serves as a base. These reactions convert readily available substrates, such as quinacridone, pentacenedione and pentacenetetraone, into conjugated skeletons, yielding several narrow-band-emissive molecules, including an actively pursued deep-blue emitter with peak emission at 450 nm and a narrow emission band of 13 nm. The tetrafold C–H activation of pentacenetetraone, with each step achieving an average yield of 95%, underscores the efficacy of iron catalysis in selectively activating C–H bonds on π-extended systems, offering promise for expanding organic electronic materials. Metal–π interactions typically hinder metal-catalysed C–H functionalization reactions involving large π-conjugated systems. Now, an iron-catalysed aza-annulation method, employing a bulky trisphosphine ligand and an aluminium base, proves efficient for large π-extended substrates, holding promise for electronic materials discovery.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


