Christopher L. Burcham*, Michael F. Doherty, Baron G. Peters, Sarah L. Price, Matteo Salvalaglio, Susan M. Reutzel-Edens, Louise S. Price, Ravi Kumar Reddy Addula, Nicholas Francia, Vikram Khanna and Yongsheng Zhao,
{"title":"制药数字化设计:从化学结构到晶体多态性再到概念结晶过程","authors":"Christopher L. Burcham*, Michael F. Doherty, Baron G. Peters, Sarah L. Price, Matteo Salvalaglio, Susan M. Reutzel-Edens, Louise S. Price, Ravi Kumar Reddy Addula, Nicholas Francia, Vikram Khanna and Yongsheng Zhao, ","doi":"10.1021/acs.cgd.3c01390","DOIUrl":null,"url":null,"abstract":"<p >A workflow for the digital design of crystallization processes starting from the chemical structure of the active pharmaceutical ingredient (API) is a multistep, multidisciplinary process. A simple version would be to first predict the API crystal structure and, from it, the corresponding properties of solubility, morphology, and growth rates, assuming that the nucleation would be controlled by seeding, and then use these parameters to design the crystallization process. This is usually an oversimplification as most APIs are polymorphic, and the most stable crystal of the API alone may not have the required properties for development into a drug product. This perspective, from the experience of a Lilly Digital Design project, considers the fundamental theoretical basis of crystal structure prediction (CSP), free energy, solubility, morphology, and growth rate prediction, and the current state of nucleation simulation. This is illustrated by applying the modeling techniques to real examples, olanzapine and succinic acid. We demonstrate the promise of using ab initio computer modeling for solid form selection and process design in pharmaceutical development. We also identify open problems in the application of current computational modeling and achieving the accuracy required for immediate implementation that currently limit the applicability of the approach.</p><p >This work considers the theoretical basis of crystal structure prediction (CSP), free energy, solubility, morphology, and growth rate prediction, and the current state of nucleation simulation to provide the conceptual process design for industrial crystallizations of pharmaceutical compounds. This is illustrated by applying the modeling techniques to real examples, olanzapine and succinic acid. We describe and demonstrate the promise of using ab initio computer modeling for solid form selection and process design in pharmaceutical development from only a molecular structure.</p>","PeriodicalId":34,"journal":{"name":"Crystal Growth & Design","volume":null,"pages":null},"PeriodicalIF":3.2000,"publicationDate":"2024-06-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.acs.org/doi/epdf/10.1021/acs.cgd.3c01390","citationCount":"0","resultStr":"{\"title\":\"Pharmaceutical Digital Design: From Chemical Structure through Crystal Polymorph to Conceptual Crystallization Process\",\"authors\":\"Christopher L. Burcham*, Michael F. Doherty, Baron G. Peters, Sarah L. Price, Matteo Salvalaglio, Susan M. Reutzel-Edens, Louise S. Price, Ravi Kumar Reddy Addula, Nicholas Francia, Vikram Khanna and Yongsheng Zhao, \",\"doi\":\"10.1021/acs.cgd.3c01390\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >A workflow for the digital design of crystallization processes starting from the chemical structure of the active pharmaceutical ingredient (API) is a multistep, multidisciplinary process. A simple version would be to first predict the API crystal structure and, from it, the corresponding properties of solubility, morphology, and growth rates, assuming that the nucleation would be controlled by seeding, and then use these parameters to design the crystallization process. This is usually an oversimplification as most APIs are polymorphic, and the most stable crystal of the API alone may not have the required properties for development into a drug product. This perspective, from the experience of a Lilly Digital Design project, considers the fundamental theoretical basis of crystal structure prediction (CSP), free energy, solubility, morphology, and growth rate prediction, and the current state of nucleation simulation. This is illustrated by applying the modeling techniques to real examples, olanzapine and succinic acid. We demonstrate the promise of using ab initio computer modeling for solid form selection and process design in pharmaceutical development. We also identify open problems in the application of current computational modeling and achieving the accuracy required for immediate implementation that currently limit the applicability of the approach.</p><p >This work considers the theoretical basis of crystal structure prediction (CSP), free energy, solubility, morphology, and growth rate prediction, and the current state of nucleation simulation to provide the conceptual process design for industrial crystallizations of pharmaceutical compounds. This is illustrated by applying the modeling techniques to real examples, olanzapine and succinic acid. We describe and demonstrate the promise of using ab initio computer modeling for solid form selection and process design in pharmaceutical development from only a molecular structure.</p>\",\"PeriodicalId\":34,\"journal\":{\"name\":\"Crystal Growth & Design\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":3.2000,\"publicationDate\":\"2024-06-24\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.acs.org/doi/epdf/10.1021/acs.cgd.3c01390\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Crystal Growth & Design\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.cgd.3c01390\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Crystal Growth & Design","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.cgd.3c01390","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
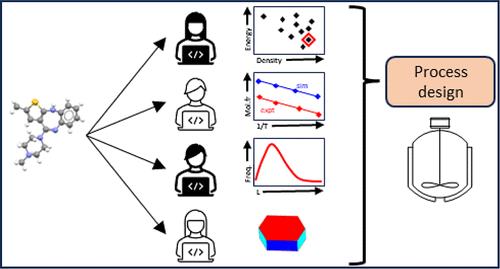
Pharmaceutical Digital Design: From Chemical Structure through Crystal Polymorph to Conceptual Crystallization Process
A workflow for the digital design of crystallization processes starting from the chemical structure of the active pharmaceutical ingredient (API) is a multistep, multidisciplinary process. A simple version would be to first predict the API crystal structure and, from it, the corresponding properties of solubility, morphology, and growth rates, assuming that the nucleation would be controlled by seeding, and then use these parameters to design the crystallization process. This is usually an oversimplification as most APIs are polymorphic, and the most stable crystal of the API alone may not have the required properties for development into a drug product. This perspective, from the experience of a Lilly Digital Design project, considers the fundamental theoretical basis of crystal structure prediction (CSP), free energy, solubility, morphology, and growth rate prediction, and the current state of nucleation simulation. This is illustrated by applying the modeling techniques to real examples, olanzapine and succinic acid. We demonstrate the promise of using ab initio computer modeling for solid form selection and process design in pharmaceutical development. We also identify open problems in the application of current computational modeling and achieving the accuracy required for immediate implementation that currently limit the applicability of the approach.
This work considers the theoretical basis of crystal structure prediction (CSP), free energy, solubility, morphology, and growth rate prediction, and the current state of nucleation simulation to provide the conceptual process design for industrial crystallizations of pharmaceutical compounds. This is illustrated by applying the modeling techniques to real examples, olanzapine and succinic acid. We describe and demonstrate the promise of using ab initio computer modeling for solid form selection and process design in pharmaceutical development from only a molecular structure.
期刊介绍:
The aim of Crystal Growth & Design is to stimulate crossfertilization of knowledge among scientists and engineers working in the fields of crystal growth, crystal engineering, and the industrial application of crystalline materials.
Crystal Growth & Design publishes theoretical and experimental studies of the physical, chemical, and biological phenomena and processes related to the design, growth, and application of crystalline materials. Synergistic approaches originating from different disciplines and technologies and integrating the fields of crystal growth, crystal engineering, intermolecular interactions, and industrial application are encouraged.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


