{"title":"石墨烯上烷烃 2D 单层形成的量子化学建模","authors":"E.S. Kartashynska","doi":"10.1016/j.jciso.2024.100117","DOIUrl":null,"url":null,"abstract":"<div><p>The paper presents a quantum chemical approach for assessment of the thermodynamic parameters of association for alkanes C<sub>n</sub>H<sub>2n+2</sub> (<em>n</em> = 6–14) and polyaromatic hydrocarbons (PAH) of the coronene series as model structures of the graphene surface within the framework of semiempirical methods. The enthalpy, entropy and Gibbs energy of formation and binding for alkanes with PAH were calculated using the PM3 and PM6-DH2 methods. It is shown that an adequate description of the interactions in the regarded complexes requires the use of PM6-DH2 method, since it contains corrections for dispersion interactions and hydrogen bonds. The parallel orientation of the alkane molecule relative to the coronene plane is proved to be more energetically preferable than perpendicular one, which is consistent with experimental data.</p><p>Intermolecular C–H/π interactions are revealed to be crucial in the 2D film formation of alkanes on graphene/graphite. While interactions between alkane molecules make a destabilizing contribution due to the implementation of energetically unfavorable types of intermolecular CH/HC interactions. This stipulates a threshold chain length of alkanes capable of film formation on the graphene/graphite surface at standard temperature: 14 and 19 carbon atoms for parallel and perpendicular oriented alkanes, respectively. The obtained threshold values of the alkane chain length, as well as the geometric parameters of their orientation in 2D monolayers on the graphene/graphite surface are consistent with available experimental data.</p></div>","PeriodicalId":73541,"journal":{"name":"JCIS open","volume":"15 ","pages":"Article 100117"},"PeriodicalIF":0.0000,"publicationDate":"2024-06-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S2666934X24000175/pdfft?md5=25768e49b6e5612608243f5b20cd1eb7&pid=1-s2.0-S2666934X24000175-main.pdf","citationCount":"0","resultStr":"{\"title\":\"Quantum chemical modeling of alkane2D monolayer formation on graphene\",\"authors\":\"E.S. Kartashynska\",\"doi\":\"10.1016/j.jciso.2024.100117\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>The paper presents a quantum chemical approach for assessment of the thermodynamic parameters of association for alkanes C<sub>n</sub>H<sub>2n+2</sub> (<em>n</em> = 6–14) and polyaromatic hydrocarbons (PAH) of the coronene series as model structures of the graphene surface within the framework of semiempirical methods. The enthalpy, entropy and Gibbs energy of formation and binding for alkanes with PAH were calculated using the PM3 and PM6-DH2 methods. It is shown that an adequate description of the interactions in the regarded complexes requires the use of PM6-DH2 method, since it contains corrections for dispersion interactions and hydrogen bonds. The parallel orientation of the alkane molecule relative to the coronene plane is proved to be more energetically preferable than perpendicular one, which is consistent with experimental data.</p><p>Intermolecular C–H/π interactions are revealed to be crucial in the 2D film formation of alkanes on graphene/graphite. While interactions between alkane molecules make a destabilizing contribution due to the implementation of energetically unfavorable types of intermolecular CH/HC interactions. This stipulates a threshold chain length of alkanes capable of film formation on the graphene/graphite surface at standard temperature: 14 and 19 carbon atoms for parallel and perpendicular oriented alkanes, respectively. The obtained threshold values of the alkane chain length, as well as the geometric parameters of their orientation in 2D monolayers on the graphene/graphite surface are consistent with available experimental data.</p></div>\",\"PeriodicalId\":73541,\"journal\":{\"name\":\"JCIS open\",\"volume\":\"15 \",\"pages\":\"Article 100117\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2024-06-27\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.sciencedirect.com/science/article/pii/S2666934X24000175/pdfft?md5=25768e49b6e5612608243f5b20cd1eb7&pid=1-s2.0-S2666934X24000175-main.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"JCIS open\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S2666934X24000175\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"Materials Science\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"JCIS open","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2666934X24000175","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"Materials Science","Score":null,"Total":0}
Quantum chemical modeling of alkane2D monolayer formation on graphene
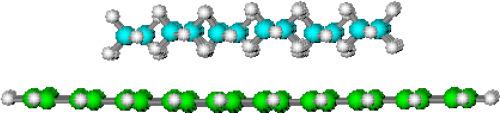
The paper presents a quantum chemical approach for assessment of the thermodynamic parameters of association for alkanes CnH2n+2 (n = 6–14) and polyaromatic hydrocarbons (PAH) of the coronene series as model structures of the graphene surface within the framework of semiempirical methods. The enthalpy, entropy and Gibbs energy of formation and binding for alkanes with PAH were calculated using the PM3 and PM6-DH2 methods. It is shown that an adequate description of the interactions in the regarded complexes requires the use of PM6-DH2 method, since it contains corrections for dispersion interactions and hydrogen bonds. The parallel orientation of the alkane molecule relative to the coronene plane is proved to be more energetically preferable than perpendicular one, which is consistent with experimental data.
Intermolecular C–H/π interactions are revealed to be crucial in the 2D film formation of alkanes on graphene/graphite. While interactions between alkane molecules make a destabilizing contribution due to the implementation of energetically unfavorable types of intermolecular CH/HC interactions. This stipulates a threshold chain length of alkanes capable of film formation on the graphene/graphite surface at standard temperature: 14 and 19 carbon atoms for parallel and perpendicular oriented alkanes, respectively. The obtained threshold values of the alkane chain length, as well as the geometric parameters of their orientation in 2D monolayers on the graphene/graphite surface are consistent with available experimental data.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


