Ryan M Riley, Gian Luca Negri, S-W Grace Cheng, Sandra E Spencer Miko, Ryan D Morin, Gregg B Morin
{"title":"针对 TMT11 和 TMT16 标记样品的质谱采集和分馏建议。","authors":"Ryan M Riley, Gian Luca Negri, S-W Grace Cheng, Sandra E Spencer Miko, Ryan D Morin, Gregg B Morin","doi":"10.1021/acs.jproteome.4c00014","DOIUrl":null,"url":null,"abstract":"<p><p>Proteome coverage and accurate protein quantification are both important for evaluating biological systems; however, compromises between quantification, coverage, and mass spectrometry (MS) resources are often necessary. Consequently, experimental parameters that impact coverage and quantification must be adjusted, depending on experimental goals. Among these parameters is offline prefractionation, which is utilized in MS-based proteomics to decrease sample complexity resulting in higher overall proteome coverage upon MS analysis. Prefractionation leads to increases in required MS analysis time, although this is often mitigated by isobaric labeling using tandem-mass tags (TMT), which allow samples to be multiplexed. Here we evaluate common prefractionation schemes, TMT variants, and MS acquisition methods and their impact on protein quantification and coverage. Furthermore, we provide recommendations for experimental design depending on the experimental goals.</p>","PeriodicalId":48,"journal":{"name":"Journal of Proteome Research","volume":null,"pages":null},"PeriodicalIF":3.8000,"publicationDate":"2024-08-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Mass Spectrometry Acquisition and Fractionation Recommendations for TMT11 and TMT16 Labeled Samples.\",\"authors\":\"Ryan M Riley, Gian Luca Negri, S-W Grace Cheng, Sandra E Spencer Miko, Ryan D Morin, Gregg B Morin\",\"doi\":\"10.1021/acs.jproteome.4c00014\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Proteome coverage and accurate protein quantification are both important for evaluating biological systems; however, compromises between quantification, coverage, and mass spectrometry (MS) resources are often necessary. Consequently, experimental parameters that impact coverage and quantification must be adjusted, depending on experimental goals. Among these parameters is offline prefractionation, which is utilized in MS-based proteomics to decrease sample complexity resulting in higher overall proteome coverage upon MS analysis. Prefractionation leads to increases in required MS analysis time, although this is often mitigated by isobaric labeling using tandem-mass tags (TMT), which allow samples to be multiplexed. Here we evaluate common prefractionation schemes, TMT variants, and MS acquisition methods and their impact on protein quantification and coverage. Furthermore, we provide recommendations for experimental design depending on the experimental goals.</p>\",\"PeriodicalId\":48,\"journal\":{\"name\":\"Journal of Proteome Research\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":3.8000,\"publicationDate\":\"2024-08-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Proteome Research\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jproteome.4c00014\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/6/29 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Proteome Research","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1021/acs.jproteome.4c00014","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/6/29 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
摘要
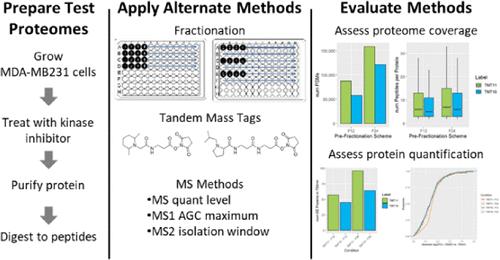
蛋白质组覆盖率和准确的蛋白质定量对于评估生物系统都很重要;然而,定量、覆盖率和质谱(MS)资源之间的折衷往往是必要的。因此,必须根据实验目标调整影响覆盖率和定量的实验参数。在这些参数中,离线预分馏是一种基于质谱的蛋白质组学方法,它可以降低样品的复杂性,从而在质谱分析时提高蛋白质组的整体覆盖率。预分馏会导致所需的质谱分析时间增加,不过使用串联质量标签(TMT)进行等位标记通常可以缓解这一问题,因为这样可以对样品进行多重分析。在此,我们对常见的预分馏方案、TMT 变体和 MS 采集方法及其对蛋白质定量和覆盖率的影响进行了评估。此外,我们还根据实验目标提供了实验设计建议。
Mass Spectrometry Acquisition and Fractionation Recommendations for TMT11 and TMT16 Labeled Samples.
Proteome coverage and accurate protein quantification are both important for evaluating biological systems; however, compromises between quantification, coverage, and mass spectrometry (MS) resources are often necessary. Consequently, experimental parameters that impact coverage and quantification must be adjusted, depending on experimental goals. Among these parameters is offline prefractionation, which is utilized in MS-based proteomics to decrease sample complexity resulting in higher overall proteome coverage upon MS analysis. Prefractionation leads to increases in required MS analysis time, although this is often mitigated by isobaric labeling using tandem-mass tags (TMT), which allow samples to be multiplexed. Here we evaluate common prefractionation schemes, TMT variants, and MS acquisition methods and their impact on protein quantification and coverage. Furthermore, we provide recommendations for experimental design depending on the experimental goals.
期刊介绍:
Journal of Proteome Research publishes content encompassing all aspects of global protein analysis and function, including the dynamic aspects of genomics, spatio-temporal proteomics, metabonomics and metabolomics, clinical and agricultural proteomics, as well as advances in methodology including bioinformatics. The theme and emphasis is on a multidisciplinary approach to the life sciences through the synergy between the different types of "omics".

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


