使用去噪自编码器的两阶段策略,实现输入基因型缺失的稳健无参考基因型归因。
IF 2.6
3区 生物学
Q2 GENETICS & HEREDITY
引用次数: 0
摘要
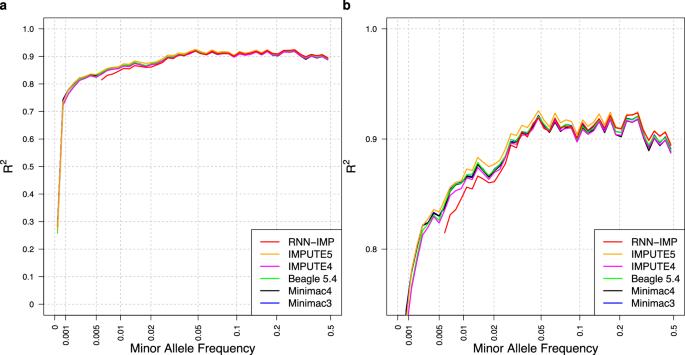
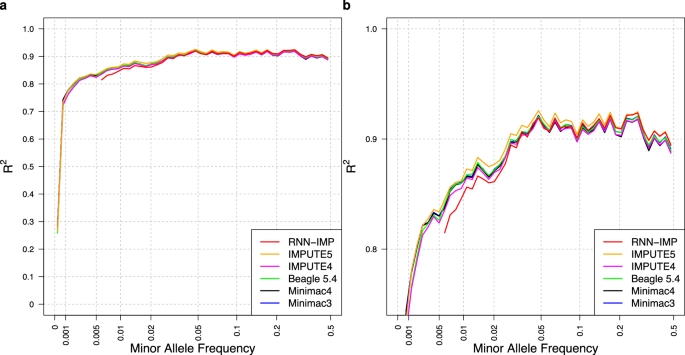
广泛使用的基因型估算方法基于李氏和斯蒂芬斯模型,该模型假定新的单倍型可以通过突变和重组修改参考面板中的现有单倍型来表示。这些方法使用 SNP 阵列中的基因型作为输入,通过分析参考面板中的重组模式来估计与输入基因型一致的单倍型,然后推断未观察到的变异。虽然这些方法需要可识别形式的参考面板,但由于隐私和同意问题,其公开使用受到限制。克服这些限制的一种策略是使用去标识化的单倍型信息,如摘要统计或模型参数。深度学习(DL)的进步为开发归因方法提供了可能,这种方法通过将单倍型信息作为模型参数处理,以无参照的方式使用单倍型信息,同时保持与基于李氏和斯蒂芬斯模型的方法相当的归因准确性。在此,我们将简要介绍基于 DL 的无参照基因型估算方法,包括我们研究小组开发的 RNN-IMP。然后,我们使用 1000 基因组计划第三阶段数据集和相应的模拟 Omni2.5 SNP 基因型数据,评估了 RNN-IMP 与广泛使用的基于 Li 和 Stephens 模型的估算方法在准确率(R2)方面的性能。虽然 RNN-IMP 对输入基因型中的缺失值很敏感,但我们提出了一种两阶段归约策略:首先使用去噪自编码器归约缺失的基因型;然后 RNN-IMP 处理这些归约的基因型。这种方法恢复了因缺失值而降低的估算精度,提高了 RNN-IMP 的实际应用。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Two-stage strategy using denoising autoencoders for robust reference-free genotype imputation with missing input genotypes
Widely used genotype imputation methods are based on the Li and Stephens model, which assumes that new haplotypes can be represented by modifying existing haplotypes in a reference panel through mutations and recombinations. These methods use genotypes from SNP arrays as inputs to estimate haplotypes that align with the input genotypes by analyzing recombination patterns within a reference panel, and then infer unobserved variants. While these methods require reference panels in an identifiable form, their public use is limited due to privacy and consent concerns. One strategy to overcome these limitations is to use de-identified haplotype information, such as summary statistics or model parameters. Advances in deep learning (DL) offer the potential to develop imputation methods that use haplotype information in a reference-free manner by handling it as model parameters, while maintaining comparable imputation accuracy to methods based on the Li and Stephens model. Here, we provide a brief introduction to DL-based reference-free genotype imputation methods, including RNN-IMP, developed by our research group. We then evaluate the performance of RNN-IMP against widely-used Li and Stephens model-based imputation methods in terms of accuracy (R2), using the 1000 Genomes Project Phase 3 dataset and corresponding simulated Omni2.5 SNP genotype data. Although RNN-IMP is sensitive to missing values in input genotypes, we propose a two-stage imputation strategy: missing genotypes are first imputed using denoising autoencoders; RNN-IMP then processes these imputed genotypes. This approach restores the imputation accuracy that is degraded by missing values, enhancing the practical use of RNN-IMP.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Journal of Human Genetics
生物-遗传学
CiteScore
7.20
自引率
0.00%
发文量
101
审稿时长
4-8 weeks
期刊介绍:
The Journal of Human Genetics is an international journal publishing articles on human genetics, including medical genetics and human genome analysis. It covers all aspects of human genetics, including molecular genetics, clinical genetics, behavioral genetics, immunogenetics, pharmacogenomics, population genetics, functional genomics, epigenetics, genetic counseling and gene therapy.
Articles on the following areas are especially welcome: genetic factors of monogenic and complex disorders, genome-wide association studies, genetic epidemiology, cancer genetics, personal genomics, genotype-phenotype relationships and genome diversity.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


