Feng Zhou, Haolin Du, Yang Wang, Weiqiang Fu, Bingchen Zhao, Jielong Zhou* and Yingsheng J. Zhang*,
{"title":"利用全原子分子动力学和机器学习解密 CBL-B 抑制剂的选择性","authors":"Feng Zhou, Haolin Du, Yang Wang, Weiqiang Fu, Bingchen Zhao, Jielong Zhou* and Yingsheng J. Zhang*, ","doi":"10.1021/acsmedchemlett.4c00047","DOIUrl":null,"url":null,"abstract":"<p >We employ a combination of accelerated molecular dynamics and machine learning to unravel how the dynamic characteristics of CBL-B and C–CBL confer their binding affinity and selectivity for ligands from subtle structural disparities within their binding pockets and dissociation pathways. Our predictive model of dissociation rate constants (<i>k</i><sub>off</sub>) demonstrates a moderate correlation between predicted <i>k</i><sub>off</sub> and experimental IC<sub>50</sub> values, which is consistent with experimental <i>k</i><sub>off</sub> and τ-random accelerated molecular dynamics (τRAMD) results. By employing a linear regression of dissociation trajectories, we identified key amino acids in binding pockets and along the dissociation paths responsible for activity and selectivity. These amino acids are statistically significant in achieving activity and selectivity and contribute to the primary structural discrepancies between CBL-B and C-CBL. Moreover, the binding free energies calculated from molecular mechanics with generalized Born and surface area solvation (MM/GBSA) highlight the Δ<i>G</i> difference between CBL-B and C-CBL. The <i>k</i><sub>off</sub> prediction, together with the key amino acids, provides important guides for designing drugs with high selectivity.</p>","PeriodicalId":20,"journal":{"name":"ACS Medicinal Chemistry Letters","volume":null,"pages":null},"PeriodicalIF":3.5000,"publicationDate":"2024-06-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Deciphering the Selectivity of CBL-B Inhibitors Using All-Atom Molecular Dynamics and Machine Learning\",\"authors\":\"Feng Zhou, Haolin Du, Yang Wang, Weiqiang Fu, Bingchen Zhao, Jielong Zhou* and Yingsheng J. Zhang*, \",\"doi\":\"10.1021/acsmedchemlett.4c00047\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >We employ a combination of accelerated molecular dynamics and machine learning to unravel how the dynamic characteristics of CBL-B and C–CBL confer their binding affinity and selectivity for ligands from subtle structural disparities within their binding pockets and dissociation pathways. Our predictive model of dissociation rate constants (<i>k</i><sub>off</sub>) demonstrates a moderate correlation between predicted <i>k</i><sub>off</sub> and experimental IC<sub>50</sub> values, which is consistent with experimental <i>k</i><sub>off</sub> and τ-random accelerated molecular dynamics (τRAMD) results. By employing a linear regression of dissociation trajectories, we identified key amino acids in binding pockets and along the dissociation paths responsible for activity and selectivity. These amino acids are statistically significant in achieving activity and selectivity and contribute to the primary structural discrepancies between CBL-B and C-CBL. Moreover, the binding free energies calculated from molecular mechanics with generalized Born and surface area solvation (MM/GBSA) highlight the Δ<i>G</i> difference between CBL-B and C-CBL. The <i>k</i><sub>off</sub> prediction, together with the key amino acids, provides important guides for designing drugs with high selectivity.</p>\",\"PeriodicalId\":20,\"journal\":{\"name\":\"ACS Medicinal Chemistry Letters\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":3.5000,\"publicationDate\":\"2024-06-05\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"ACS Medicinal Chemistry Letters\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acsmedchemlett.4c00047\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS Medicinal Chemistry Letters","FirstCategoryId":"3","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acsmedchemlett.4c00047","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
Deciphering the Selectivity of CBL-B Inhibitors Using All-Atom Molecular Dynamics and Machine Learning
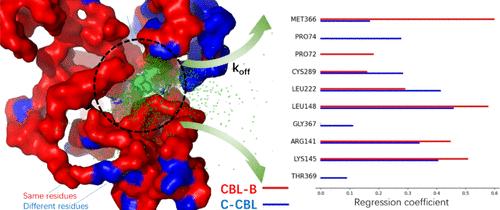
We employ a combination of accelerated molecular dynamics and machine learning to unravel how the dynamic characteristics of CBL-B and C–CBL confer their binding affinity and selectivity for ligands from subtle structural disparities within their binding pockets and dissociation pathways. Our predictive model of dissociation rate constants (koff) demonstrates a moderate correlation between predicted koff and experimental IC50 values, which is consistent with experimental koff and τ-random accelerated molecular dynamics (τRAMD) results. By employing a linear regression of dissociation trajectories, we identified key amino acids in binding pockets and along the dissociation paths responsible for activity and selectivity. These amino acids are statistically significant in achieving activity and selectivity and contribute to the primary structural discrepancies between CBL-B and C-CBL. Moreover, the binding free energies calculated from molecular mechanics with generalized Born and surface area solvation (MM/GBSA) highlight the ΔG difference between CBL-B and C-CBL. The koff prediction, together with the key amino acids, provides important guides for designing drugs with high selectivity.
期刊介绍:
ACS Medicinal Chemistry Letters is interested in receiving manuscripts that discuss various aspects of medicinal chemistry. The journal will publish studies that pertain to a broad range of subject matter, including compound design and optimization, biological evaluation, drug delivery, imaging agents, and pharmacology of both small and large bioactive molecules. Specific areas include but are not limited to:
Identification, synthesis, and optimization of lead biologically active molecules and drugs (small molecules and biologics)
Biological characterization of new molecular entities in the context of drug discovery
Computational, cheminformatics, and structural studies for the identification or SAR analysis of bioactive molecules, ligands and their targets, etc.
Novel and improved methodologies, including radiation biochemistry, with broad application to medicinal chemistry
Discovery technologies for biologically active molecules from both synthetic and natural (plant and other) sources
Pharmacokinetic/pharmacodynamic studies that address mechanisms underlying drug disposition and response
Pharmacogenetic and pharmacogenomic studies used to enhance drug design and the translation of medicinal chemistry into the clinic
Mechanistic drug metabolism and regulation of metabolic enzyme gene expression
Chemistry patents relevant to the medicinal chemistry field.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


