基因组时代的新一代数据过滤
IF 39.1
1区 生物学
Q1 GENETICS & HEREDITY
引用次数: 0
摘要
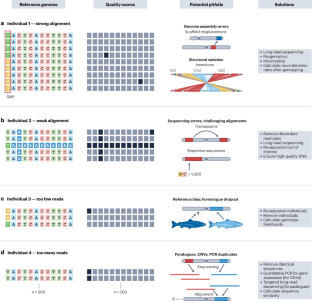
基因组数据在各学科中无处不在,从农业到生物多样性、生态学、进化论和人类健康。然而,这些数据集往往包含噪音或错误,而且缺少信息,会影响后续计算分析和结论的准确性和可靠性。基因组数据分析的一个关键步骤是过滤--从数据集中移除测序碱基、读数、基因变异和/或个体--以提高下游分析的数据质量。在过滤基因组数据时,研究人员面临着多种选择;他们必须选择应用哪些过滤器,并选择适当的阈值。为了帮助开创下一代基因组数据过滤技术,我们回顾并提出了最佳实践,以改进基因组数据集常用过滤类型和阈值的实施、可重复性和报告标准。我们主要关注小等位基因频率、每个个体或每个位点的缺失数据、连锁不平衡和哈代-温伯格偏差的过滤。我们利用模拟和经验数据集说明了不同过滤阈值对常见种群遗传学统计的巨大影响,如田岛 D 值、种群分化 (FST)、核苷酸多样性 (π) 和有效种群规模 (Ne)。本文章由计算机程序翻译,如有差异,请以英文原文为准。

Next-generation data filtering in the genomics era
Genomic data are ubiquitous across disciplines, from agriculture to biodiversity, ecology, evolution and human health. However, these datasets often contain noise or errors and are missing information that can affect the accuracy and reliability of subsequent computational analyses and conclusions. A key step in genomic data analysis is filtering — removing sequencing bases, reads, genetic variants and/or individuals from a dataset — to improve data quality for downstream analyses. Researchers are confronted with a multitude of choices when filtering genomic data; they must choose which filters to apply and select appropriate thresholds. To help usher in the next generation of genomic data filtering, we review and suggest best practices to improve the implementation, reproducibility and reporting standards for filter types and thresholds commonly applied to genomic datasets. We focus mainly on filters for minor allele frequency, missing data per individual or per locus, linkage disequilibrium and Hardy–Weinberg deviations. Using simulated and empirical datasets, we illustrate the large effects of different filtering thresholds on common population genetics statistics, such as Tajima’s D value, population differentiation (FST), nucleotide diversity (π) and effective population size (Ne). Filtering genomic data is a crucial step to ensure the quality and reliability of downstream analyses. The authors provide guidance on the choice of filtering strategies and thresholds, including filters that remove sequencing bases or reads, variants, loci, genotypes or individuals from genomic datasets to improve accuracy and reproducibility.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊
Nature Reviews Genetics
生物-遗传学
CiteScore
57.40
自引率
0.50%
发文量
113
审稿时长
6-12 weeks
期刊介绍:
At Nature Reviews Genetics, our goal is to be the leading source of reviews and commentaries for the scientific communities we serve. We are dedicated to publishing authoritative articles that are easily accessible to our readers. We believe in enhancing our articles with clear and understandable figures, tables, and other display items. Our aim is to provide an unparalleled service to authors, referees, and readers, and we are committed to maximizing the usefulness and impact of each article we publish.
Within our journal, we publish a range of content including Research Highlights, Comments, Reviews, and Perspectives that are relevant to geneticists and genomicists. With our broad scope, we ensure that the articles we publish reach the widest possible audience.
As part of the Nature Reviews portfolio of journals, we strive to uphold the high standards and reputation associated with this esteemed collection of publications.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


