Khaja Shameem Mohammed Abdul , Kimin Han , Alyssa B. Guerrero , Cekia N. Wilson , Amogh Kulkarni , Nicole H. Purcell
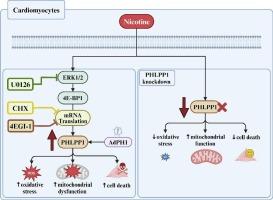
{"title":"通过 ERK-4E-BP1 信号轴增加 PHLPP1 的表达驱动尼古丁诱导的氧化应激相关心肌细胞损伤。","authors":"Khaja Shameem Mohammed Abdul , Kimin Han , Alyssa B. Guerrero , Cekia N. Wilson , Amogh Kulkarni , Nicole H. Purcell","doi":"10.1016/j.yjmcc.2024.05.014","DOIUrl":null,"url":null,"abstract":"<div><p>Nicotine, a key constituent of tobacco/electronic cigarettes causes cardiovascular injury and mortality. Nicotine is known to induce oxidative stress and mitochondrial dysfunction in cardiomyocytes leading to cell death. However, the underlying mechanisms remain unclear. Pleckstrin homology domain leucine-rich repeat protein phosphatase (PHLPP) is a member of metal-dependent protein phosphatase (PPM) family and is known to dephosphorylate several AGC family kinases and thereby regulate a diverse set of cellular functions including cell growth, survival, and death. Our lab has previously demonstrated that PHLPP1 removal reduced cardiomyocyte death and cardiac dysfunction following injury. Here, we present a novel finding that nicotine exposure significantly increased PHLPP1 protein expression in the adolescent rodent heart. Building upon our in vivo finding, we determined the mechanism of PHLPP1 expression in cardiomyocytes. Nicotine significantly increased PHLPP1 protein expression without altering PHLPP2 in cardiomyocytes. In cardiomyocytes, nicotine significantly increased NADPH oxidase 4 (NOX4), which coincided with increased reactive oxygen species (ROS) and increased cardiomyocyte apoptosis which were dependent on PHLPP1 expression. PHLPP1 expression was both necessary and sufficient for nicotine induced mitochondrial dysfunction. Mechanistically, nicotine activated extracellular signal-regulated protein kinases (ERK1/2) and subsequent eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1) to increase PHLPP1 protein expression. Inhibition of protein synthesis with cycloheximide (CHX) and 4EGI-1 abolished nicotine induced PHLPP1 protein expression. Moreover, inhibition of ERK1/2 activity by U0126 significantly blocked nicotine induced PHLPP1 expression. Overall, this study reveals a novel mechanism by which nicotine regulates PHLPP1 expression through ERK-4E-BP1 signaling axis to drive cardiomyocyte injury.</p></div>","PeriodicalId":16402,"journal":{"name":"Journal of molecular and cellular cardiology","volume":"193 ","pages":"Pages 100-112"},"PeriodicalIF":4.9000,"publicationDate":"2024-06-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S0022282824000889/pdfft?md5=b4f171c91334adb9e3bc6443798ece68&pid=1-s2.0-S0022282824000889-main.pdf","citationCount":"0","resultStr":"{\"title\":\"Increased PHLPP1 expression through ERK-4E-BP1 signaling axis drives nicotine induced oxidative stress related damage of cardiomyocytes\",\"authors\":\"Khaja Shameem Mohammed Abdul , Kimin Han , Alyssa B. Guerrero , Cekia N. Wilson , Amogh Kulkarni , Nicole H. Purcell\",\"doi\":\"10.1016/j.yjmcc.2024.05.014\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Nicotine, a key constituent of tobacco/electronic cigarettes causes cardiovascular injury and mortality. Nicotine is known to induce oxidative stress and mitochondrial dysfunction in cardiomyocytes leading to cell death. However, the underlying mechanisms remain unclear. Pleckstrin homology domain leucine-rich repeat protein phosphatase (PHLPP) is a member of metal-dependent protein phosphatase (PPM) family and is known to dephosphorylate several AGC family kinases and thereby regulate a diverse set of cellular functions including cell growth, survival, and death. Our lab has previously demonstrated that PHLPP1 removal reduced cardiomyocyte death and cardiac dysfunction following injury. Here, we present a novel finding that nicotine exposure significantly increased PHLPP1 protein expression in the adolescent rodent heart. Building upon our in vivo finding, we determined the mechanism of PHLPP1 expression in cardiomyocytes. Nicotine significantly increased PHLPP1 protein expression without altering PHLPP2 in cardiomyocytes. In cardiomyocytes, nicotine significantly increased NADPH oxidase 4 (NOX4), which coincided with increased reactive oxygen species (ROS) and increased cardiomyocyte apoptosis which were dependent on PHLPP1 expression. PHLPP1 expression was both necessary and sufficient for nicotine induced mitochondrial dysfunction. Mechanistically, nicotine activated extracellular signal-regulated protein kinases (ERK1/2) and subsequent eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1) to increase PHLPP1 protein expression. Inhibition of protein synthesis with cycloheximide (CHX) and 4EGI-1 abolished nicotine induced PHLPP1 protein expression. Moreover, inhibition of ERK1/2 activity by U0126 significantly blocked nicotine induced PHLPP1 expression. Overall, this study reveals a novel mechanism by which nicotine regulates PHLPP1 expression through ERK-4E-BP1 signaling axis to drive cardiomyocyte injury.</p></div>\",\"PeriodicalId\":16402,\"journal\":{\"name\":\"Journal of molecular and cellular cardiology\",\"volume\":\"193 \",\"pages\":\"Pages 100-112\"},\"PeriodicalIF\":4.9000,\"publicationDate\":\"2024-06-06\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.sciencedirect.com/science/article/pii/S0022282824000889/pdfft?md5=b4f171c91334adb9e3bc6443798ece68&pid=1-s2.0-S0022282824000889-main.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of molecular and cellular cardiology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S0022282824000889\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CARDIAC & CARDIOVASCULAR SYSTEMS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of molecular and cellular cardiology","FirstCategoryId":"3","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0022282824000889","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CARDIAC & CARDIOVASCULAR SYSTEMS","Score":null,"Total":0}
Increased PHLPP1 expression through ERK-4E-BP1 signaling axis drives nicotine induced oxidative stress related damage of cardiomyocytes
Nicotine, a key constituent of tobacco/electronic cigarettes causes cardiovascular injury and mortality. Nicotine is known to induce oxidative stress and mitochondrial dysfunction in cardiomyocytes leading to cell death. However, the underlying mechanisms remain unclear. Pleckstrin homology domain leucine-rich repeat protein phosphatase (PHLPP) is a member of metal-dependent protein phosphatase (PPM) family and is known to dephosphorylate several AGC family kinases and thereby regulate a diverse set of cellular functions including cell growth, survival, and death. Our lab has previously demonstrated that PHLPP1 removal reduced cardiomyocyte death and cardiac dysfunction following injury. Here, we present a novel finding that nicotine exposure significantly increased PHLPP1 protein expression in the adolescent rodent heart. Building upon our in vivo finding, we determined the mechanism of PHLPP1 expression in cardiomyocytes. Nicotine significantly increased PHLPP1 protein expression without altering PHLPP2 in cardiomyocytes. In cardiomyocytes, nicotine significantly increased NADPH oxidase 4 (NOX4), which coincided with increased reactive oxygen species (ROS) and increased cardiomyocyte apoptosis which were dependent on PHLPP1 expression. PHLPP1 expression was both necessary and sufficient for nicotine induced mitochondrial dysfunction. Mechanistically, nicotine activated extracellular signal-regulated protein kinases (ERK1/2) and subsequent eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1) to increase PHLPP1 protein expression. Inhibition of protein synthesis with cycloheximide (CHX) and 4EGI-1 abolished nicotine induced PHLPP1 protein expression. Moreover, inhibition of ERK1/2 activity by U0126 significantly blocked nicotine induced PHLPP1 expression. Overall, this study reveals a novel mechanism by which nicotine regulates PHLPP1 expression through ERK-4E-BP1 signaling axis to drive cardiomyocyte injury.
期刊介绍:
The Journal of Molecular and Cellular Cardiology publishes work advancing knowledge of the mechanisms responsible for both normal and diseased cardiovascular function. To this end papers are published in all relevant areas. These include (but are not limited to): structural biology; genetics; proteomics; morphology; stem cells; molecular biology; metabolism; biophysics; bioengineering; computational modeling and systems analysis; electrophysiology; pharmacology and physiology. Papers are encouraged with both basic and translational approaches. The journal is directed not only to basic scientists but also to clinical cardiologists who wish to follow the rapidly advancing frontiers of basic knowledge of the heart and circulation.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


