{"title":"从城市污水中分离出的柔性志贺氏菌的比较基因组学及其特征。","authors":"Sarmishta Mukhopadhyay, Meesha Singh, Mahashweta Mitra Ghosh, Santanu Chakrabarti, Sayak Ganguli","doi":"10.1264/jsme2.ME23105","DOIUrl":null,"url":null,"abstract":"<p><p>Shigella species are a group of highly transmissible Gram-negative pathogens. Increasing reports of infection with extensively drug-resistant varieties of this stomach bug has convinced the World Health Organization to prioritize Shigella for novel therapeutic interventions. We herein coupled the whole-genome sequencing of a natural isolate of Shigella flexneri with a pangenome ana-lysis to characterize pathogen genomics within this species, which will provide us with an insight into its existing genomic diversity and highlight the root causes behind the emergence of quick vaccine escape variants. The isolated novel strain of S. flexneri contained ~4,500 protein-coding genes, 57 of which imparted resistance to antibiotics. A comparative pan-genomic ana-lysis revealed genomic variability of ~64%, the shared conservation of core genes in central metabolic processes, and the enrichment of unique/accessory genes in virulence and defense mechanisms that contributed to much of the observed antimicrobial resistance (AMR). A pathway ana-lysis of the core genome mapped 22 genes to 2 antimicrobial resistance pathways, with the bulk coding for multidrug efflux pumps and two component regulatory systems that are considered to work synergistically towards the development of resistance phenotypes. The prospective evolvability of Shigella species as witnessed by the marked difference in genomic content, the strain-specific essentiality of unique/accessory genes, and the inclusion of a potent resistance mechanism within the core genome, strengthens the possibility of novel serotypes emerging in the near future and emphasizes the importance of tracking down genomic diversity in drug/vaccine design and AMR governance.</p>","PeriodicalId":18482,"journal":{"name":"Microbes and Environments","volume":"39 2","pages":""},"PeriodicalIF":2.0000,"publicationDate":"2024-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11220449/pdf/","citationCount":"0","resultStr":"{\"title\":\"Comparative Genomics and Characterization of Shigella flexneri Isolated from Urban Wastewater.\",\"authors\":\"Sarmishta Mukhopadhyay, Meesha Singh, Mahashweta Mitra Ghosh, Santanu Chakrabarti, Sayak Ganguli\",\"doi\":\"10.1264/jsme2.ME23105\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Shigella species are a group of highly transmissible Gram-negative pathogens. Increasing reports of infection with extensively drug-resistant varieties of this stomach bug has convinced the World Health Organization to prioritize Shigella for novel therapeutic interventions. We herein coupled the whole-genome sequencing of a natural isolate of Shigella flexneri with a pangenome ana-lysis to characterize pathogen genomics within this species, which will provide us with an insight into its existing genomic diversity and highlight the root causes behind the emergence of quick vaccine escape variants. The isolated novel strain of S. flexneri contained ~4,500 protein-coding genes, 57 of which imparted resistance to antibiotics. A comparative pan-genomic ana-lysis revealed genomic variability of ~64%, the shared conservation of core genes in central metabolic processes, and the enrichment of unique/accessory genes in virulence and defense mechanisms that contributed to much of the observed antimicrobial resistance (AMR). A pathway ana-lysis of the core genome mapped 22 genes to 2 antimicrobial resistance pathways, with the bulk coding for multidrug efflux pumps and two component regulatory systems that are considered to work synergistically towards the development of resistance phenotypes. The prospective evolvability of Shigella species as witnessed by the marked difference in genomic content, the strain-specific essentiality of unique/accessory genes, and the inclusion of a potent resistance mechanism within the core genome, strengthens the possibility of novel serotypes emerging in the near future and emphasizes the importance of tracking down genomic diversity in drug/vaccine design and AMR governance.</p>\",\"PeriodicalId\":18482,\"journal\":{\"name\":\"Microbes and Environments\",\"volume\":\"39 2\",\"pages\":\"\"},\"PeriodicalIF\":2.0000,\"publicationDate\":\"2024-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11220449/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Microbes and Environments\",\"FirstCategoryId\":\"93\",\"ListUrlMain\":\"https://doi.org/10.1264/jsme2.ME23105\",\"RegionNum\":4,\"RegionCategory\":\"环境科学与生态学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"BIOTECHNOLOGY & APPLIED MICROBIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Microbes and Environments","FirstCategoryId":"93","ListUrlMain":"https://doi.org/10.1264/jsme2.ME23105","RegionNum":4,"RegionCategory":"环境科学与生态学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOTECHNOLOGY & APPLIED MICROBIOLOGY","Score":null,"Total":0}
Comparative Genomics and Characterization of Shigella flexneri Isolated from Urban Wastewater.
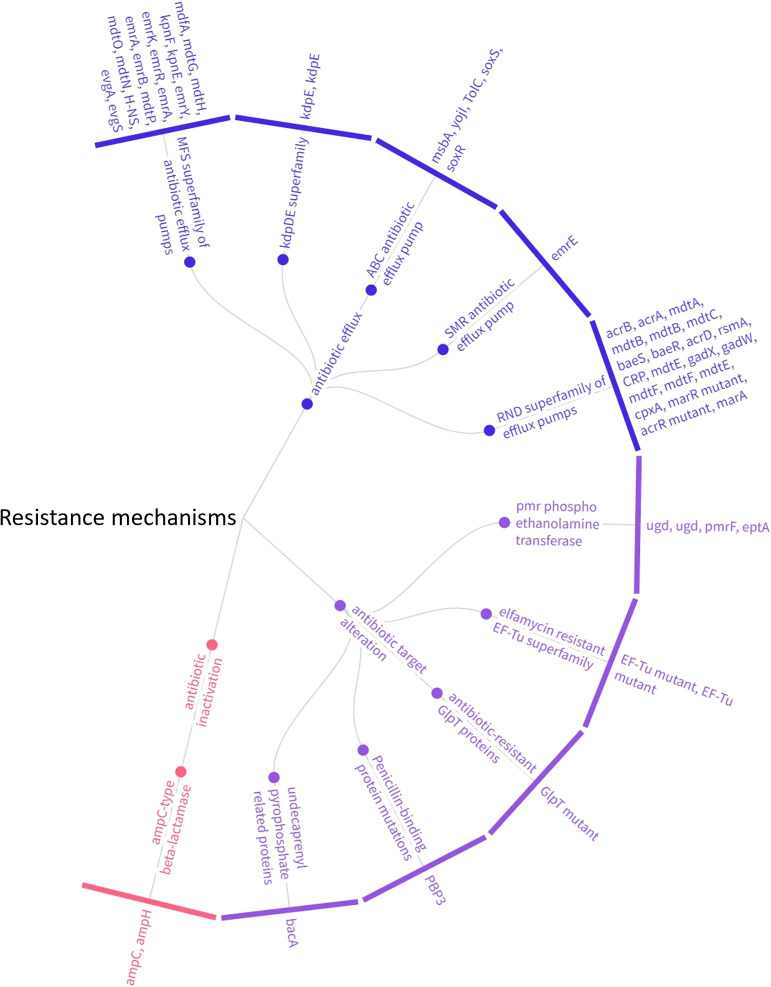
Shigella species are a group of highly transmissible Gram-negative pathogens. Increasing reports of infection with extensively drug-resistant varieties of this stomach bug has convinced the World Health Organization to prioritize Shigella for novel therapeutic interventions. We herein coupled the whole-genome sequencing of a natural isolate of Shigella flexneri with a pangenome ana-lysis to characterize pathogen genomics within this species, which will provide us with an insight into its existing genomic diversity and highlight the root causes behind the emergence of quick vaccine escape variants. The isolated novel strain of S. flexneri contained ~4,500 protein-coding genes, 57 of which imparted resistance to antibiotics. A comparative pan-genomic ana-lysis revealed genomic variability of ~64%, the shared conservation of core genes in central metabolic processes, and the enrichment of unique/accessory genes in virulence and defense mechanisms that contributed to much of the observed antimicrobial resistance (AMR). A pathway ana-lysis of the core genome mapped 22 genes to 2 antimicrobial resistance pathways, with the bulk coding for multidrug efflux pumps and two component regulatory systems that are considered to work synergistically towards the development of resistance phenotypes. The prospective evolvability of Shigella species as witnessed by the marked difference in genomic content, the strain-specific essentiality of unique/accessory genes, and the inclusion of a potent resistance mechanism within the core genome, strengthens the possibility of novel serotypes emerging in the near future and emphasizes the importance of tracking down genomic diversity in drug/vaccine design and AMR governance.
期刊介绍:
Microbial ecology in natural and engineered environments; Microbial degradation of xenobiotic compounds; Microbial processes in biogeochemical cycles; Microbial interactions and signaling with animals and plants; Interactions among microorganisms; Microorganisms related to public health; Phylogenetic and functional diversity of microbial communities; Genomics, metagenomics, and bioinformatics for microbiology; Application of microorganisms to agriculture, fishery, and industry; Molecular biology and biochemistry related to environmental microbiology; Methodology in general and environmental microbiology; Interdisciplinary research areas for microbial ecology (e.g., Astrobiology, and Origins of Life); Taxonomic description of novel microorganisms with ecological perspective; Physiology and metabolisms of microorganisms; Evolution of genes and microorganisms; Genome report of microorganisms with ecological perspective.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


