Amanda Momenzadeh, Simion Kreimer, Dongchuan Guo, Matthew Ayres, Daniel Berman, Kuang-Yuh Chyu, Prediman K Shah, Dianna Milewicz, Ali Azizzadeh, Jesse G Meyer, Sarah Parker
{"title":"利用机器学习和血浆蛋白质组特征区分降主动脉疾病","authors":"Amanda Momenzadeh, Simion Kreimer, Dongchuan Guo, Matthew Ayres, Daniel Berman, Kuang-Yuh Chyu, Prediman K Shah, Dianna Milewicz, Ali Azizzadeh, Jesse G Meyer, Sarah Parker","doi":"10.1186/s12014-024-09487-4","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Descending thoracic aortic aneurysms and dissections can go undetected until severe and catastrophic, and few clinical indices exist to screen for aneurysms or predict risk of dissection.</p><p><strong>Methods: </strong>This study generated a plasma proteomic dataset from 75 patients with descending type B dissection (Type B) and 62 patients with descending thoracic aortic aneurysm (DTAA). Standard statistical approaches were compared to supervised machine learning (ML) algorithms to distinguish Type B from DTAA cases. Quantitatively similar proteins were clustered based on linkage distance from hierarchical clustering and ML models were trained with uncorrelated protein lists across various linkage distances with hyperparameter optimization using fivefold cross validation. Permutation importance (PI) was used for ranking the most important predictor proteins of ML classification between disease states and the proteins among the top 10 PI protein groups were submitted for pathway analysis.</p><p><strong>Results: </strong>Of the 1,549 peptides and 198 proteins used in this study, no peptides and only one protein, hemopexin (HPX), were significantly different at an adjusted p < 0.01 between Type B and DTAA cases. The highest performing model on the training set (Support Vector Classifier) and its corresponding linkage distance (0.5) were used for evaluation of the test set, yielding a precision-recall area under the curve of 0.7 to classify between Type B from DTAA cases. The five proteins with the highest PI scores were immunoglobulin heavy variable 6-1 (IGHV6-1), lecithin-cholesterol acyltransferase (LCAT), coagulation factor 12 (F12), HPX, and immunoglobulin heavy variable 4-4 (IGHV4-4). All proteins from the top 10 most important groups generated the following significantly enriched pathways in the plasma of Type B versus DTAA patients: complement activation, humoral immune response, and blood coagulation.</p><p><strong>Conclusions: </strong>We conclude that ML may be useful in differentiating the plasma proteome of highly similar disease states that would otherwise not be distinguishable using statistics, and, in such cases, ML may enable prioritizing important proteins for model prediction.</p>","PeriodicalId":10468,"journal":{"name":"Clinical proteomics","volume":"21 1","pages":"38"},"PeriodicalIF":3.3000,"publicationDate":"2024-06-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11145886/pdf/","citationCount":"0","resultStr":"{\"title\":\"Differentiation between descending thoracic aortic diseases using machine learning and plasma proteomic signatures.\",\"authors\":\"Amanda Momenzadeh, Simion Kreimer, Dongchuan Guo, Matthew Ayres, Daniel Berman, Kuang-Yuh Chyu, Prediman K Shah, Dianna Milewicz, Ali Azizzadeh, Jesse G Meyer, Sarah Parker\",\"doi\":\"10.1186/s12014-024-09487-4\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Descending thoracic aortic aneurysms and dissections can go undetected until severe and catastrophic, and few clinical indices exist to screen for aneurysms or predict risk of dissection.</p><p><strong>Methods: </strong>This study generated a plasma proteomic dataset from 75 patients with descending type B dissection (Type B) and 62 patients with descending thoracic aortic aneurysm (DTAA). Standard statistical approaches were compared to supervised machine learning (ML) algorithms to distinguish Type B from DTAA cases. Quantitatively similar proteins were clustered based on linkage distance from hierarchical clustering and ML models were trained with uncorrelated protein lists across various linkage distances with hyperparameter optimization using fivefold cross validation. Permutation importance (PI) was used for ranking the most important predictor proteins of ML classification between disease states and the proteins among the top 10 PI protein groups were submitted for pathway analysis.</p><p><strong>Results: </strong>Of the 1,549 peptides and 198 proteins used in this study, no peptides and only one protein, hemopexin (HPX), were significantly different at an adjusted p < 0.01 between Type B and DTAA cases. The highest performing model on the training set (Support Vector Classifier) and its corresponding linkage distance (0.5) were used for evaluation of the test set, yielding a precision-recall area under the curve of 0.7 to classify between Type B from DTAA cases. The five proteins with the highest PI scores were immunoglobulin heavy variable 6-1 (IGHV6-1), lecithin-cholesterol acyltransferase (LCAT), coagulation factor 12 (F12), HPX, and immunoglobulin heavy variable 4-4 (IGHV4-4). All proteins from the top 10 most important groups generated the following significantly enriched pathways in the plasma of Type B versus DTAA patients: complement activation, humoral immune response, and blood coagulation.</p><p><strong>Conclusions: </strong>We conclude that ML may be useful in differentiating the plasma proteome of highly similar disease states that would otherwise not be distinguishable using statistics, and, in such cases, ML may enable prioritizing important proteins for model prediction.</p>\",\"PeriodicalId\":10468,\"journal\":{\"name\":\"Clinical proteomics\",\"volume\":\"21 1\",\"pages\":\"38\"},\"PeriodicalIF\":3.3000,\"publicationDate\":\"2024-06-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11145886/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Clinical proteomics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s12014-024-09487-4\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical proteomics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s12014-024-09487-4","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
摘要
背景:降主动脉瘤和主动脉夹层可能在严重和灾难性之前一直未被发现,几乎没有临床指标可用于筛查动脉瘤或预测夹层风险:本研究从 75 例降支 B 型夹层(B 型)患者和 62 例降支胸主动脉瘤(DTAA)患者中生成了血浆蛋白质组数据集。将标准统计方法与有监督的机器学习(ML)算法进行了比较,以区分 B 型和 DTAA 病例。根据分层聚类的链接距离对定量相似的蛋白质进行聚类,并使用五倍交叉验证的超参数优化方法,在不同链接距离的非相关蛋白质列表中训练 ML 模型。采用置换重要性(PI)对疾病状态之间的 ML 分类中最重要的预测蛋白质进行排序,并将 PI 排名前 10 位的蛋白质组提交进行通路分析:结果:在本研究使用的 1,549 种肽和 198 种蛋白质中,没有肽和一种蛋白质(血卟啉(HPX))在调整后的 p 值上有显著差异:我们得出结论:ML 可能有助于区分高度相似的疾病状态的血浆蛋白质组,否则用统计学方法是无法区分的,在这种情况下,ML 可能有助于优先选择重要蛋白质进行模型预测。
Differentiation between descending thoracic aortic diseases using machine learning and plasma proteomic signatures.
Background: Descending thoracic aortic aneurysms and dissections can go undetected until severe and catastrophic, and few clinical indices exist to screen for aneurysms or predict risk of dissection.
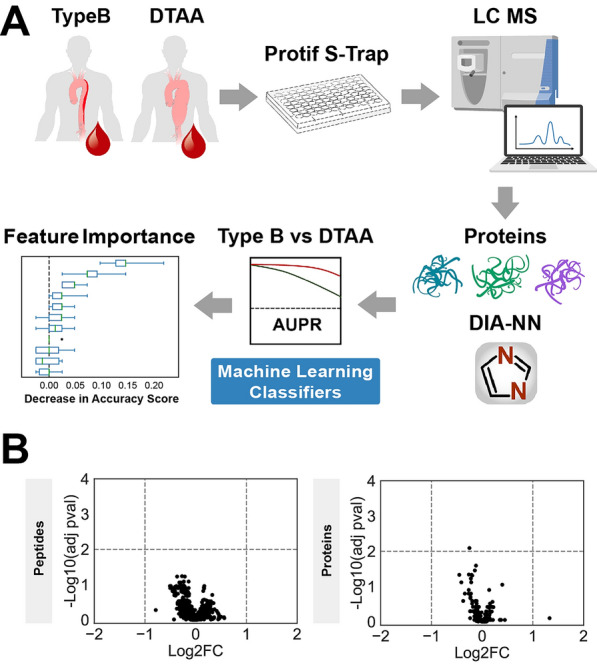
Methods: This study generated a plasma proteomic dataset from 75 patients with descending type B dissection (Type B) and 62 patients with descending thoracic aortic aneurysm (DTAA). Standard statistical approaches were compared to supervised machine learning (ML) algorithms to distinguish Type B from DTAA cases. Quantitatively similar proteins were clustered based on linkage distance from hierarchical clustering and ML models were trained with uncorrelated protein lists across various linkage distances with hyperparameter optimization using fivefold cross validation. Permutation importance (PI) was used for ranking the most important predictor proteins of ML classification between disease states and the proteins among the top 10 PI protein groups were submitted for pathway analysis.
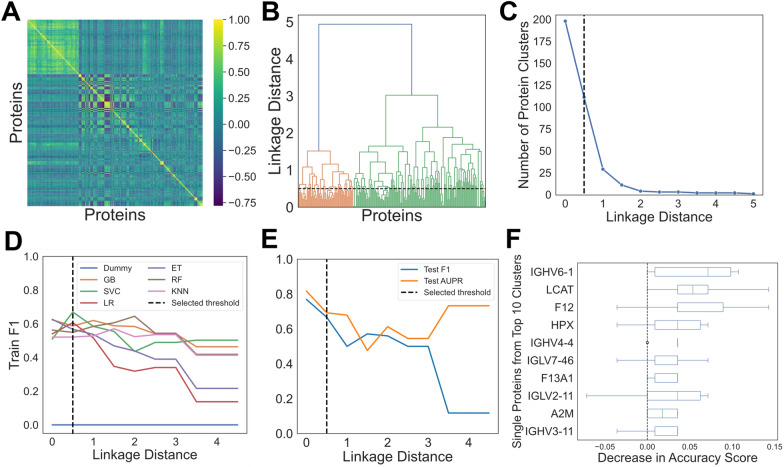
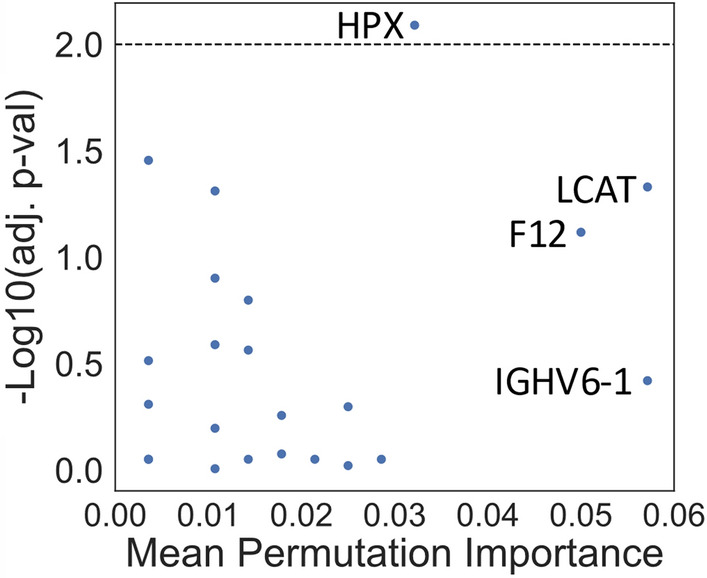
Results: Of the 1,549 peptides and 198 proteins used in this study, no peptides and only one protein, hemopexin (HPX), were significantly different at an adjusted p < 0.01 between Type B and DTAA cases. The highest performing model on the training set (Support Vector Classifier) and its corresponding linkage distance (0.5) were used for evaluation of the test set, yielding a precision-recall area under the curve of 0.7 to classify between Type B from DTAA cases. The five proteins with the highest PI scores were immunoglobulin heavy variable 6-1 (IGHV6-1), lecithin-cholesterol acyltransferase (LCAT), coagulation factor 12 (F12), HPX, and immunoglobulin heavy variable 4-4 (IGHV4-4). All proteins from the top 10 most important groups generated the following significantly enriched pathways in the plasma of Type B versus DTAA patients: complement activation, humoral immune response, and blood coagulation.
Conclusions: We conclude that ML may be useful in differentiating the plasma proteome of highly similar disease states that would otherwise not be distinguishable using statistics, and, in such cases, ML may enable prioritizing important proteins for model prediction.
期刊介绍:
Clinical Proteomics encompasses all aspects of translational proteomics. Special emphasis will be placed on the application of proteomic technology to all aspects of clinical research and molecular medicine. The journal is committed to rapid scientific review and timely publication of submitted manuscripts.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


