{"title":"用机器学习预测单粒子密度矩阵","authors":"S. Hazra, U. Patil and S. Sanvito*, ","doi":"10.1021/acs.jctc.4c00042","DOIUrl":null,"url":null,"abstract":"<p >Two of the most widely used electronic-structure theory methods, namely, Hartree–Fock and Kohn–Sham density functional theory, require the iterative solution of a set of Schrödinger-like equations. The speed of convergence of such a process depends on the complexity of the system under investigation, the self-consistent-field algorithm employed, and the initial guess for the density matrix. An initial density matrix close to the ground-state matrix will effectively allow one to cut out many of the self-consistent steps necessary to achieve convergence. Here, we predict the density matrix of Kohn–Sham density functional theory by constructing a neural network that uses only the atomic positions as information. Such a neural network provides an initial guess for the density matrix far superior to that of any other recipes available. Furthermore, the quality of such a neural-network density matrix is good enough for the evaluation of interatomic forces. This allows us to run accelerated ab initio molecular dynamics with little to no self-consistent steps.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"20 11","pages":"4569–4578"},"PeriodicalIF":5.5000,"publicationDate":"2024-05-31","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.acs.org/doi/epdf/10.1021/acs.jctc.4c00042","citationCount":"0","resultStr":"{\"title\":\"Predicting the One-Particle Density Matrix with Machine Learning\",\"authors\":\"S. Hazra, U. Patil and S. Sanvito*, \",\"doi\":\"10.1021/acs.jctc.4c00042\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Two of the most widely used electronic-structure theory methods, namely, Hartree–Fock and Kohn–Sham density functional theory, require the iterative solution of a set of Schrödinger-like equations. The speed of convergence of such a process depends on the complexity of the system under investigation, the self-consistent-field algorithm employed, and the initial guess for the density matrix. An initial density matrix close to the ground-state matrix will effectively allow one to cut out many of the self-consistent steps necessary to achieve convergence. Here, we predict the density matrix of Kohn–Sham density functional theory by constructing a neural network that uses only the atomic positions as information. Such a neural network provides an initial guess for the density matrix far superior to that of any other recipes available. Furthermore, the quality of such a neural-network density matrix is good enough for the evaluation of interatomic forces. This allows us to run accelerated ab initio molecular dynamics with little to no self-consistent steps.</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\"20 11\",\"pages\":\"4569–4578\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2024-05-31\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.acs.org/doi/epdf/10.1021/acs.jctc.4c00042\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jctc.4c00042\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.4c00042","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Predicting the One-Particle Density Matrix with Machine Learning
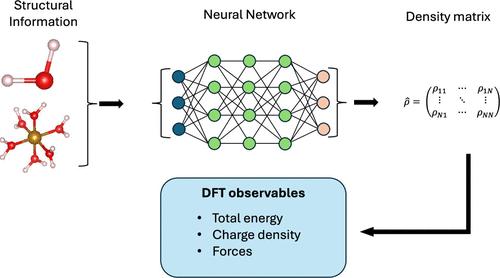
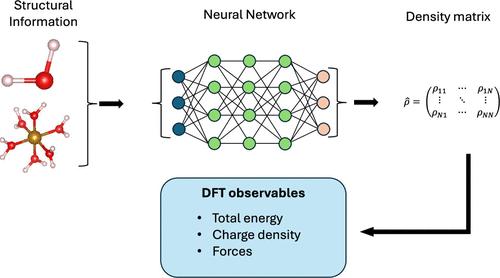
Two of the most widely used electronic-structure theory methods, namely, Hartree–Fock and Kohn–Sham density functional theory, require the iterative solution of a set of Schrödinger-like equations. The speed of convergence of such a process depends on the complexity of the system under investigation, the self-consistent-field algorithm employed, and the initial guess for the density matrix. An initial density matrix close to the ground-state matrix will effectively allow one to cut out many of the self-consistent steps necessary to achieve convergence. Here, we predict the density matrix of Kohn–Sham density functional theory by constructing a neural network that uses only the atomic positions as information. Such a neural network provides an initial guess for the density matrix far superior to that of any other recipes available. Furthermore, the quality of such a neural-network density matrix is good enough for the evaluation of interatomic forces. This allows us to run accelerated ab initio molecular dynamics with little to no self-consistent steps.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


