Adriana Amaral Carvalho, Renato Assis Machado, Célia Márcia Fernandes Maia, Luis Antônio Nogueira Dos Santos, Daniella Reis Barbosa Martelli, Ricardo Della Coletta, Hercílio Martelli Júnior
{"title":"一种罕见的 LMNA 错义突变导致下颌骨骶骨发育不良 A 型的严重表型:病例报告。","authors":"Adriana Amaral Carvalho, Renato Assis Machado, Célia Márcia Fernandes Maia, Luis Antônio Nogueira Dos Santos, Daniella Reis Barbosa Martelli, Ricardo Della Coletta, Hercílio Martelli Júnior","doi":"10.1590/1984-0462/2024/42/2022189","DOIUrl":null,"url":null,"abstract":"<p><strong>Objective: </strong>To report the case of a girl presenting a severe phenotype of mandibuloacral dysplasia type A (MADA) characterized by prominent osteolytic changes and ectodermal defects, associated with a rare homozygous LMNA missense mutation (c.1579C>T).</p><p><strong>Case description: </strong>A 6-year-old girl was evaluated during hospitalization exhibiting the following dysmorphic signs: subtotal alopecia, dysmorphic facies with prominent eyes, marked micrognathia and retrognathia, small beaked nose, teeth crowding and thin lips, generalized lipodystrophy, narrow and sloping shoulders, generalized joint stiffness and bone reabsorption in the terminal phalanges. In dermatological examination, atrophic skin, loss of cutaneous elasticity, hyperkeratosis, dermal calcinosis, and hyperpigmented and hypochromic patches were observed. Radiology exams performed showed bilateral absence of the mandibular condyles, clavicle resorption with local amorphous bone mass confluence with the scapulae, shoulder joints with subluxation and severe bone dysplasia, hip dysplasia, osteopenia and subcutaneous calcifications.</p><p><strong>Comments: </strong>MADA is a rare autosomal recessive disease caused by mutations in LMNA gene. It is characterized by craniofacial deformities, skeletal anomalies, skin alterations, lipodystrophy in certain regions of the body and premature ageing. Typical MADA is caused by the p.R527H mutation in the LMNA gene. However, molecular analysis performed from oral epithelial cells obtained from the patient showed the rare mutation c.1579C>T, p. R527C in the exon 9 of LMNA. This is the sixth family identified with this mutation described in the literature.</p>","PeriodicalId":74721,"journal":{"name":"Revista paulista de pediatria : orgao oficial da Sociedade de Pediatria de Sao Paulo","volume":"42 ","pages":"e2022189"},"PeriodicalIF":2.0000,"publicationDate":"2024-05-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11135898/pdf/","citationCount":"0","resultStr":"{\"title\":\"A rare LMNA missense mutation causing a severe phenotype of mandibuloacral dysplasia type A: a case report.\",\"authors\":\"Adriana Amaral Carvalho, Renato Assis Machado, Célia Márcia Fernandes Maia, Luis Antônio Nogueira Dos Santos, Daniella Reis Barbosa Martelli, Ricardo Della Coletta, Hercílio Martelli Júnior\",\"doi\":\"10.1590/1984-0462/2024/42/2022189\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Objective: </strong>To report the case of a girl presenting a severe phenotype of mandibuloacral dysplasia type A (MADA) characterized by prominent osteolytic changes and ectodermal defects, associated with a rare homozygous LMNA missense mutation (c.1579C>T).</p><p><strong>Case description: </strong>A 6-year-old girl was evaluated during hospitalization exhibiting the following dysmorphic signs: subtotal alopecia, dysmorphic facies with prominent eyes, marked micrognathia and retrognathia, small beaked nose, teeth crowding and thin lips, generalized lipodystrophy, narrow and sloping shoulders, generalized joint stiffness and bone reabsorption in the terminal phalanges. In dermatological examination, atrophic skin, loss of cutaneous elasticity, hyperkeratosis, dermal calcinosis, and hyperpigmented and hypochromic patches were observed. Radiology exams performed showed bilateral absence of the mandibular condyles, clavicle resorption with local amorphous bone mass confluence with the scapulae, shoulder joints with subluxation and severe bone dysplasia, hip dysplasia, osteopenia and subcutaneous calcifications.</p><p><strong>Comments: </strong>MADA is a rare autosomal recessive disease caused by mutations in LMNA gene. It is characterized by craniofacial deformities, skeletal anomalies, skin alterations, lipodystrophy in certain regions of the body and premature ageing. Typical MADA is caused by the p.R527H mutation in the LMNA gene. However, molecular analysis performed from oral epithelial cells obtained from the patient showed the rare mutation c.1579C>T, p. R527C in the exon 9 of LMNA. This is the sixth family identified with this mutation described in the literature.</p>\",\"PeriodicalId\":74721,\"journal\":{\"name\":\"Revista paulista de pediatria : orgao oficial da Sociedade de Pediatria de Sao Paulo\",\"volume\":\"42 \",\"pages\":\"e2022189\"},\"PeriodicalIF\":2.0000,\"publicationDate\":\"2024-05-27\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11135898/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Revista paulista de pediatria : orgao oficial da Sociedade de Pediatria de Sao Paulo\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1590/1984-0462/2024/42/2022189\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Revista paulista de pediatria : orgao oficial da Sociedade de Pediatria de Sao Paulo","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1590/1984-0462/2024/42/2022189","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
摘要
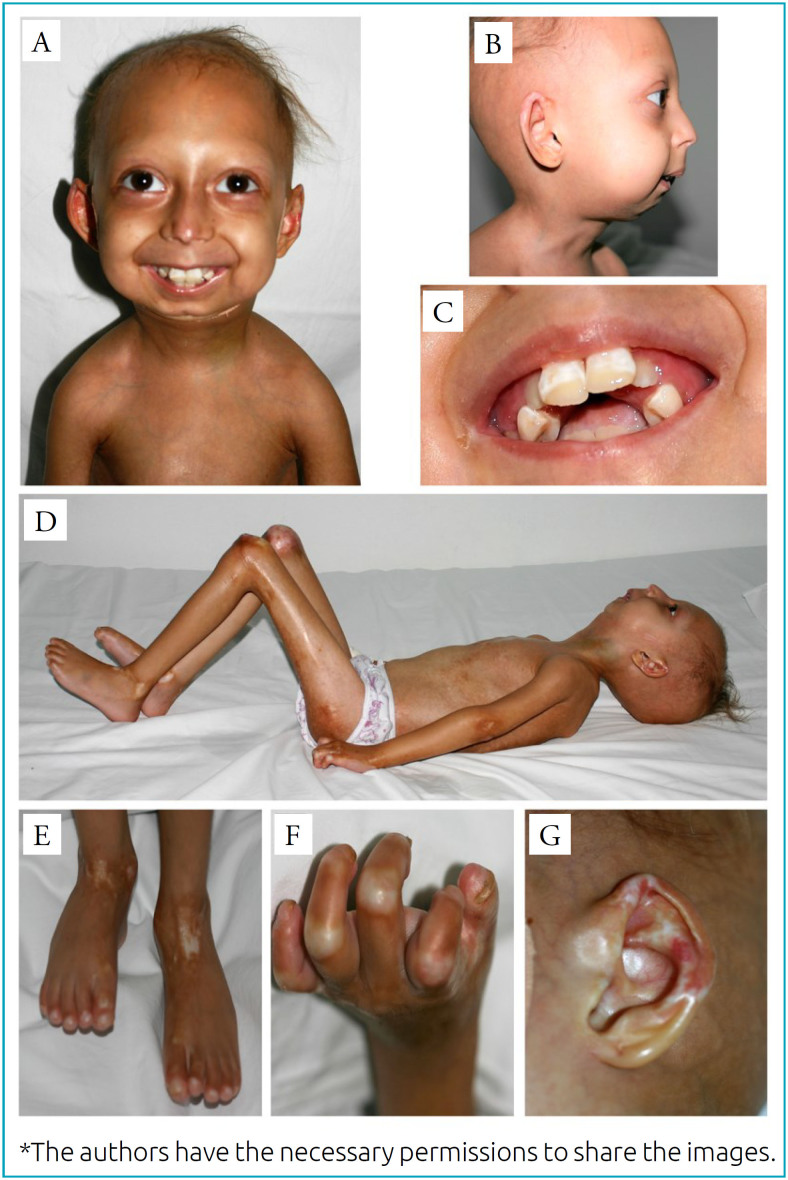
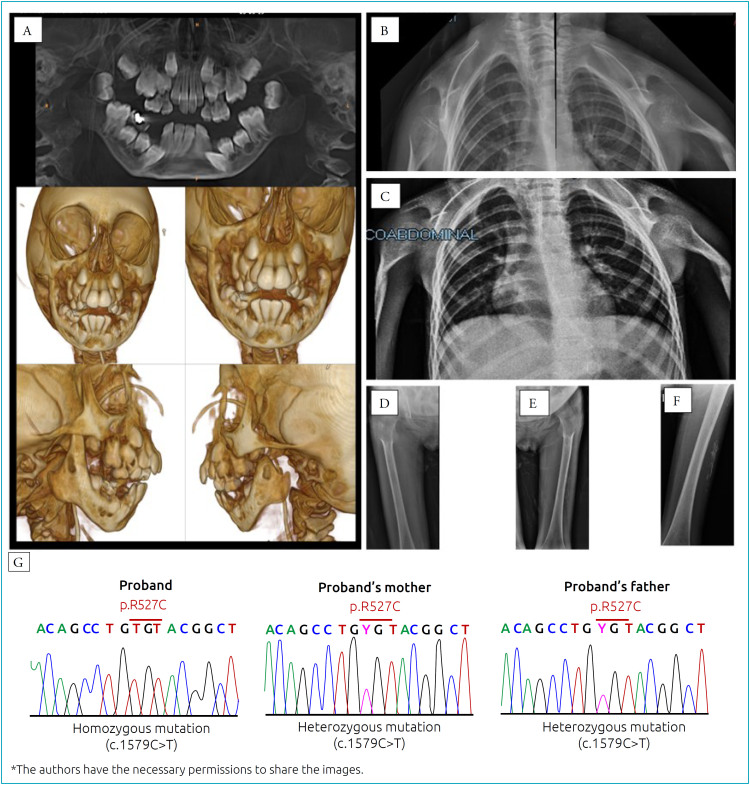
目的报告一例严重下颌骨骶骨发育不良 A 型(MADA)女孩的病例,该病例以突出的溶骨性改变和外胚层缺陷为特征,与罕见的同基因 LMNA 错义突变(c.1579C>T)有关:一名 6 岁女孩在住院期间接受了评估,发现她有以下畸形体征:次全脱发、畸形面容(眼球突出)、明显的小颌畸形和后颌畸形、小喙鼻、牙齿拥挤和薄嘴唇、全身性唇营养不良、肩部狭窄和倾斜、全身性关节僵硬和末节趾骨骨吸收。在皮肤科检查中,观察到皮肤萎缩、皮肤失去弹性、角化过度、真皮钙化以及色素沉着和色素减退斑块。放射学检查显示双侧下颌骨髁状突缺失,锁骨吸收,局部无定形骨块与肩胛骨汇合,肩关节半脱位和严重骨发育不良,髋关节发育不良,骨质疏松和皮下钙化:MADA是一种罕见的常染色体隐性遗传病,由LMNA基因突变引起。其特征为颅面畸形、骨骼异常、皮肤改变、身体某些部位脂肪营养不良和过早衰老。典型的 MADA 是由 LMNA 基因 p.R527H 突变引起的。然而,对患者口腔上皮细胞进行的分子分析表明,LMNA 第 9 外显子中存在罕见的 c.1579C>T、p.R527C 突变。这是文献中描述的第六个出现这种突变的家族。
A rare LMNA missense mutation causing a severe phenotype of mandibuloacral dysplasia type A: a case report.
Objective: To report the case of a girl presenting a severe phenotype of mandibuloacral dysplasia type A (MADA) characterized by prominent osteolytic changes and ectodermal defects, associated with a rare homozygous LMNA missense mutation (c.1579C>T).
Case description: A 6-year-old girl was evaluated during hospitalization exhibiting the following dysmorphic signs: subtotal alopecia, dysmorphic facies with prominent eyes, marked micrognathia and retrognathia, small beaked nose, teeth crowding and thin lips, generalized lipodystrophy, narrow and sloping shoulders, generalized joint stiffness and bone reabsorption in the terminal phalanges. In dermatological examination, atrophic skin, loss of cutaneous elasticity, hyperkeratosis, dermal calcinosis, and hyperpigmented and hypochromic patches were observed. Radiology exams performed showed bilateral absence of the mandibular condyles, clavicle resorption with local amorphous bone mass confluence with the scapulae, shoulder joints with subluxation and severe bone dysplasia, hip dysplasia, osteopenia and subcutaneous calcifications.
Comments: MADA is a rare autosomal recessive disease caused by mutations in LMNA gene. It is characterized by craniofacial deformities, skeletal anomalies, skin alterations, lipodystrophy in certain regions of the body and premature ageing. Typical MADA is caused by the p.R527H mutation in the LMNA gene. However, molecular analysis performed from oral epithelial cells obtained from the patient showed the rare mutation c.1579C>T, p. R527C in the exon 9 of LMNA. This is the sixth family identified with this mutation described in the literature.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


