Maitray A Patel, Mark Daley, Logan R Van Nynatten, Marat Slessarev, Gediminas Cepinskas, Douglas D Fraser
{"title":"通过血浆抗体微阵列和机器学习确定 Covid-19 重症患者蛋白质组特征的减少。","authors":"Maitray A Patel, Mark Daley, Logan R Van Nynatten, Marat Slessarev, Gediminas Cepinskas, Douglas D Fraser","doi":"10.1186/s12014-024-09488-3","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>COVID-19 is a complex, multi-system disease with varying severity and symptoms. Identifying changes in critically ill COVID-19 patients' proteomes enables a better understanding of markers associated with susceptibility, symptoms, and treatment. We performed plasma antibody microarray and machine learning analyses to identify novel proteins of COVID-19.</p><p><strong>Methods: </strong>A case-control study comparing the concentration of 2000 plasma proteins in age- and sex-matched COVID-19 inpatients, non-COVID-19 sepsis controls, and healthy control subjects. Machine learning was used to identify a unique proteome signature in COVID-19 patients. Protein expression was correlated with clinically relevant variables and analyzed for temporal changes over hospitalization days 1, 3, 7, and 10. Expert-curated protein expression information was analyzed with Natural language processing (NLP) to determine organ- and cell-specific expression.</p><p><strong>Results: </strong>Machine learning identified a 28-protein model that accurately differentiated COVID-19 patients from ICU non-COVID-19 patients (accuracy = 0.89, AUC = 1.00, F1 = 0.89) and healthy controls (accuracy = 0.89, AUC = 1.00, F1 = 0.88). An optimal nine-protein model (PF4V1, NUCB1, CrkL, SerpinD1, Fen1, GATA-4, ProSAAS, PARK7, and NET1) maintained high classification ability. Specific proteins correlated with hemoglobin, coagulation factors, hypertension, and high-flow nasal cannula intervention (P < 0.01). Time-course analysis of the 28 leading proteins demonstrated no significant temporal changes within the COVID-19 cohort. NLP analysis identified multi-system expression of the key proteins, with the digestive and nervous systems being the leading systems.</p><p><strong>Conclusions: </strong>The plasma proteome of critically ill COVID-19 patients was distinguishable from that of non-COVID-19 sepsis controls and healthy control subjects. The leading 28 proteins and their subset of 9 proteins yielded accurate classification models and are expressed in multiple organ systems. The identified COVID-19 proteomic signature helps elucidate COVID-19 pathophysiology and may guide future COVID-19 treatment development.</p>","PeriodicalId":10468,"journal":{"name":"Clinical proteomics","volume":"21 1","pages":"33"},"PeriodicalIF":3.3000,"publicationDate":"2024-05-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11100131/pdf/","citationCount":"0","resultStr":"{\"title\":\"A reduced proteomic signature in critically ill Covid-19 patients determined with plasma antibody micro-array and machine learning.\",\"authors\":\"Maitray A Patel, Mark Daley, Logan R Van Nynatten, Marat Slessarev, Gediminas Cepinskas, Douglas D Fraser\",\"doi\":\"10.1186/s12014-024-09488-3\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>COVID-19 is a complex, multi-system disease with varying severity and symptoms. Identifying changes in critically ill COVID-19 patients' proteomes enables a better understanding of markers associated with susceptibility, symptoms, and treatment. We performed plasma antibody microarray and machine learning analyses to identify novel proteins of COVID-19.</p><p><strong>Methods: </strong>A case-control study comparing the concentration of 2000 plasma proteins in age- and sex-matched COVID-19 inpatients, non-COVID-19 sepsis controls, and healthy control subjects. Machine learning was used to identify a unique proteome signature in COVID-19 patients. Protein expression was correlated with clinically relevant variables and analyzed for temporal changes over hospitalization days 1, 3, 7, and 10. Expert-curated protein expression information was analyzed with Natural language processing (NLP) to determine organ- and cell-specific expression.</p><p><strong>Results: </strong>Machine learning identified a 28-protein model that accurately differentiated COVID-19 patients from ICU non-COVID-19 patients (accuracy = 0.89, AUC = 1.00, F1 = 0.89) and healthy controls (accuracy = 0.89, AUC = 1.00, F1 = 0.88). An optimal nine-protein model (PF4V1, NUCB1, CrkL, SerpinD1, Fen1, GATA-4, ProSAAS, PARK7, and NET1) maintained high classification ability. Specific proteins correlated with hemoglobin, coagulation factors, hypertension, and high-flow nasal cannula intervention (P < 0.01). Time-course analysis of the 28 leading proteins demonstrated no significant temporal changes within the COVID-19 cohort. NLP analysis identified multi-system expression of the key proteins, with the digestive and nervous systems being the leading systems.</p><p><strong>Conclusions: </strong>The plasma proteome of critically ill COVID-19 patients was distinguishable from that of non-COVID-19 sepsis controls and healthy control subjects. The leading 28 proteins and their subset of 9 proteins yielded accurate classification models and are expressed in multiple organ systems. The identified COVID-19 proteomic signature helps elucidate COVID-19 pathophysiology and may guide future COVID-19 treatment development.</p>\",\"PeriodicalId\":10468,\"journal\":{\"name\":\"Clinical proteomics\",\"volume\":\"21 1\",\"pages\":\"33\"},\"PeriodicalIF\":3.3000,\"publicationDate\":\"2024-05-17\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11100131/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Clinical proteomics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s12014-024-09488-3\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical proteomics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s12014-024-09488-3","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
A reduced proteomic signature in critically ill Covid-19 patients determined with plasma antibody micro-array and machine learning.
Background: COVID-19 is a complex, multi-system disease with varying severity and symptoms. Identifying changes in critically ill COVID-19 patients' proteomes enables a better understanding of markers associated with susceptibility, symptoms, and treatment. We performed plasma antibody microarray and machine learning analyses to identify novel proteins of COVID-19.
Methods: A case-control study comparing the concentration of 2000 plasma proteins in age- and sex-matched COVID-19 inpatients, non-COVID-19 sepsis controls, and healthy control subjects. Machine learning was used to identify a unique proteome signature in COVID-19 patients. Protein expression was correlated with clinically relevant variables and analyzed for temporal changes over hospitalization days 1, 3, 7, and 10. Expert-curated protein expression information was analyzed with Natural language processing (NLP) to determine organ- and cell-specific expression.
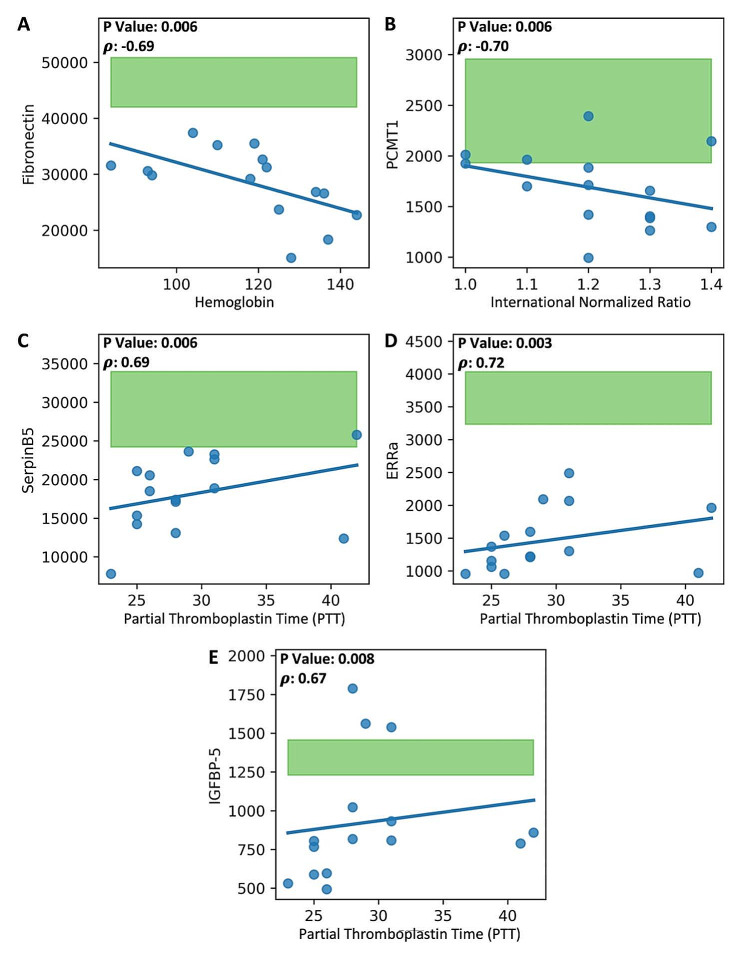
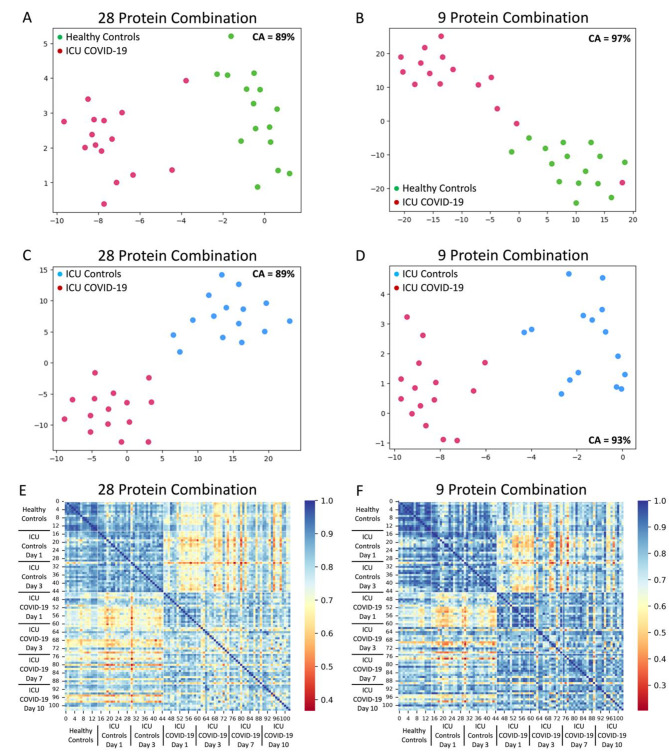
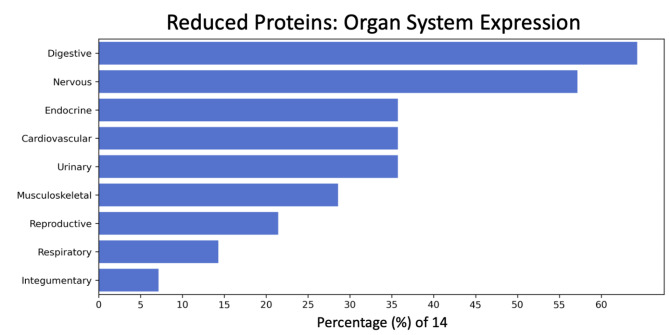
Results: Machine learning identified a 28-protein model that accurately differentiated COVID-19 patients from ICU non-COVID-19 patients (accuracy = 0.89, AUC = 1.00, F1 = 0.89) and healthy controls (accuracy = 0.89, AUC = 1.00, F1 = 0.88). An optimal nine-protein model (PF4V1, NUCB1, CrkL, SerpinD1, Fen1, GATA-4, ProSAAS, PARK7, and NET1) maintained high classification ability. Specific proteins correlated with hemoglobin, coagulation factors, hypertension, and high-flow nasal cannula intervention (P < 0.01). Time-course analysis of the 28 leading proteins demonstrated no significant temporal changes within the COVID-19 cohort. NLP analysis identified multi-system expression of the key proteins, with the digestive and nervous systems being the leading systems.
Conclusions: The plasma proteome of critically ill COVID-19 patients was distinguishable from that of non-COVID-19 sepsis controls and healthy control subjects. The leading 28 proteins and their subset of 9 proteins yielded accurate classification models and are expressed in multiple organ systems. The identified COVID-19 proteomic signature helps elucidate COVID-19 pathophysiology and may guide future COVID-19 treatment development.
期刊介绍:
Clinical Proteomics encompasses all aspects of translational proteomics. Special emphasis will be placed on the application of proteomic technology to all aspects of clinical research and molecular medicine. The journal is committed to rapid scientific review and timely publication of submitted manuscripts.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


