Roberta Russo, Achille Iolascon, Immacolata Andolfo, Roberta Marra, Barbara Eleni Rosato
{"title":"遗传性红细胞生成障碍性贫血的临床和实验室方面的最新进展。","authors":"Roberta Russo, Achille Iolascon, Immacolata Andolfo, Roberta Marra, Barbara Eleni Rosato","doi":"10.1111/ijlh.14307","DOIUrl":null,"url":null,"abstract":"<p>Hereditary dyserythropoietic anemias, or congenital dyserythropoietic anemias (CDAs), are rare disorders disrupting normal erythroid lineage development, resulting in ineffective erythropoiesis and monolinear cytopenia. CDAs include three main types (I, II, III), transcription-factor-related forms, and syndromic forms. The widespread use of next-generation sequencing in the last decade has unveiled novel causative genes and unexpected genotype–phenotype correlations. The discovery of the genetic defects underlying the CDAs not only facilitates accurate diagnosis but also enhances understanding of CDA pathophysiology. Notable advancements include identifying a hepatic-specific role of the <i>SEC23B</i> loss-of-function in iron metabolism dysregulation in CDA II, deepening <i>CDIN1</i> dysfunction during erythroid differentiation, and uncovering a recessive CDA III form associated with <i>RACGAP1</i> variants. Current treatments primarily rely on supportive measures tailored to disease severity and clinical features. Comparative studies with pyruvate kinase deficiency have illuminated new therapeutic avenues by elucidating iron dyshomeostasis and dyserythropoiesis mechanisms. We herein discuss recent progress in diagnostic methodologies, novel gene discoveries, and enhanced comprehension of CDA pathogenesis and molecular genetics.</p>","PeriodicalId":14120,"journal":{"name":"International Journal of Laboratory Hematology","volume":"46 4","pages":"595-605"},"PeriodicalIF":2.2000,"publicationDate":"2024-05-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/ijlh.14307","citationCount":"0","resultStr":"{\"title\":\"Updates on clinical and laboratory aspects of hereditary dyserythropoietic anemias\",\"authors\":\"Roberta Russo, Achille Iolascon, Immacolata Andolfo, Roberta Marra, Barbara Eleni Rosato\",\"doi\":\"10.1111/ijlh.14307\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Hereditary dyserythropoietic anemias, or congenital dyserythropoietic anemias (CDAs), are rare disorders disrupting normal erythroid lineage development, resulting in ineffective erythropoiesis and monolinear cytopenia. CDAs include three main types (I, II, III), transcription-factor-related forms, and syndromic forms. The widespread use of next-generation sequencing in the last decade has unveiled novel causative genes and unexpected genotype–phenotype correlations. The discovery of the genetic defects underlying the CDAs not only facilitates accurate diagnosis but also enhances understanding of CDA pathophysiology. Notable advancements include identifying a hepatic-specific role of the <i>SEC23B</i> loss-of-function in iron metabolism dysregulation in CDA II, deepening <i>CDIN1</i> dysfunction during erythroid differentiation, and uncovering a recessive CDA III form associated with <i>RACGAP1</i> variants. Current treatments primarily rely on supportive measures tailored to disease severity and clinical features. Comparative studies with pyruvate kinase deficiency have illuminated new therapeutic avenues by elucidating iron dyshomeostasis and dyserythropoiesis mechanisms. We herein discuss recent progress in diagnostic methodologies, novel gene discoveries, and enhanced comprehension of CDA pathogenesis and molecular genetics.</p>\",\"PeriodicalId\":14120,\"journal\":{\"name\":\"International Journal of Laboratory Hematology\",\"volume\":\"46 4\",\"pages\":\"595-605\"},\"PeriodicalIF\":2.2000,\"publicationDate\":\"2024-05-15\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1111/ijlh.14307\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"International Journal of Laboratory Hematology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1111/ijlh.14307\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"HEMATOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Journal of Laboratory Hematology","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/ijlh.14307","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"HEMATOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
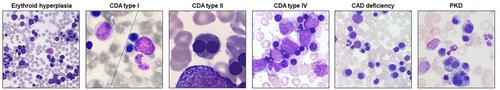
遗传性红细胞生成障碍性贫血或先天性红细胞生成障碍性贫血(CDA)是一种罕见的疾病,会破坏红细胞系的正常发育,导致红细胞生成障碍和单核细胞减少。CDA 包括三种主要类型(I、II、III)、转录因子相关类型和综合征类型。近十年来,新一代测序技术的广泛应用揭示了新的致病基因和意想不到的基因型表型相关性。发现 CDA 的基因缺陷不仅有助于准确诊断,还能加深对 CDA 病理生理学的理解。值得注意的进展包括确定了 SEC23B 功能缺失在 CDA II 铁代谢失调中的肝特异性作用,深化了 CDIN1 在红细胞分化过程中的功能障碍,并发现了与 RACGAP1 变异相关的隐性 CDA III 型。目前的治疗主要依赖于针对疾病严重程度和临床特征的支持性措施。与丙酮酸激酶缺乏症的比较研究通过阐明铁失衡和红细胞生成障碍的机制,为治疗提供了新的途径。我们在此讨论诊断方法的最新进展、新基因的发现以及对 CDA 发病机制和分子遗传学的进一步理解。
Updates on clinical and laboratory aspects of hereditary dyserythropoietic anemias
Hereditary dyserythropoietic anemias, or congenital dyserythropoietic anemias (CDAs), are rare disorders disrupting normal erythroid lineage development, resulting in ineffective erythropoiesis and monolinear cytopenia. CDAs include three main types (I, II, III), transcription-factor-related forms, and syndromic forms. The widespread use of next-generation sequencing in the last decade has unveiled novel causative genes and unexpected genotype–phenotype correlations. The discovery of the genetic defects underlying the CDAs not only facilitates accurate diagnosis but also enhances understanding of CDA pathophysiology. Notable advancements include identifying a hepatic-specific role of the SEC23B loss-of-function in iron metabolism dysregulation in CDA II, deepening CDIN1 dysfunction during erythroid differentiation, and uncovering a recessive CDA III form associated with RACGAP1 variants. Current treatments primarily rely on supportive measures tailored to disease severity and clinical features. Comparative studies with pyruvate kinase deficiency have illuminated new therapeutic avenues by elucidating iron dyshomeostasis and dyserythropoiesis mechanisms. We herein discuss recent progress in diagnostic methodologies, novel gene discoveries, and enhanced comprehension of CDA pathogenesis and molecular genetics.
期刊介绍:
The International Journal of Laboratory Hematology provides a forum for the communication of new developments, research topics and the practice of laboratory haematology.
The journal publishes invited reviews, full length original articles, and correspondence.
The International Journal of Laboratory Hematology is the official journal of the International Society for Laboratory Hematology, which addresses the following sub-disciplines: cellular analysis, flow cytometry, haemostasis and thrombosis, molecular diagnostics, haematology informatics, haemoglobinopathies, point of care testing, standards and guidelines.
The journal was launched in 2006 as the successor to Clinical and Laboratory Hematology, which was first published in 1979. An active and positive editorial policy ensures that work of a high scientific standard is reported, in order to bridge the gap between practical and academic aspects of laboratory haematology.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


