{"title":"普通白蜡树(Fraxinus excelsior L.)种群抗蚜虫基因组预测","authors":"Joanna Meger, Bartosz Ulaszewski, Małgorzata Pałucka, Czesław Kozioł, Jarosław Burczyk","doi":"10.1111/eva.13694","DOIUrl":null,"url":null,"abstract":"<p>The increase in introduced insect pests and pathogens due to anthropogenic environmental changes has become a major concern for tree species worldwide. Common ash (<i>Fraxinus excelsior</i> L.) is one of such species facing a significant threat from the invasive fungal pathogen <i>Hymenoscyphus fraxineus</i>. Some studies have indicated that the susceptibility of ash to the pathogen is genetically determined, providing some hope for accelerated breeding programs that are aimed at increasing the resistance of ash populations. To address this challenge, we used a genomic selection strategy to identify potential genetic markers that are associated with resistance to the pathogen causing ash dieback. Through genome-wide association studies (GWAS) of 300 common ash individuals from 30 populations across Poland (ddRAD, dataset A), we identified six significant SNP loci with a <i>p</i>-value ≤1 × 10<sup>−4</sup> associated with health status. To further evaluate the effectiveness of GWAS markers in predicting health status, we considered two genomic prediction scenarios. Firstly, we conducted cross-validation on dataset A. Secondly, we trained markers on dataset A and tested them on dataset B, which involved whole-genome sequencing of 20 individuals from two populations. Genomic prediction analysis revealed that the top SNPs identified via GWAS exhibited notably higher prediction accuracies compared to randomly selected SNPs, particularly with a larger number of SNPs. Cross-validation analyses using dataset A showcased high genomic prediction accuracy, predicting tree health status with over 90% accuracy across the top SNP sets ranging from 500 to 10,000 SNPs from the GWAS datasets. However, no significant results emerged for health status when the model trained on dataset A was tested on dataset B. Our findings illuminate potential genetic markers associated with resistance to ash dieback, offering support for future breeding programs in Poland aimed at combating ash dieback and bolstering conservation efforts for this invaluable tree species.</p>","PeriodicalId":168,"journal":{"name":"Evolutionary Applications","volume":null,"pages":null},"PeriodicalIF":3.5000,"publicationDate":"2024-05-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/eva.13694","citationCount":"0","resultStr":"{\"title\":\"Genomic prediction of resistance to Hymenoscyphus fraxineus in common ash (Fraxinus excelsior L.) populations\",\"authors\":\"Joanna Meger, Bartosz Ulaszewski, Małgorzata Pałucka, Czesław Kozioł, Jarosław Burczyk\",\"doi\":\"10.1111/eva.13694\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>The increase in introduced insect pests and pathogens due to anthropogenic environmental changes has become a major concern for tree species worldwide. Common ash (<i>Fraxinus excelsior</i> L.) is one of such species facing a significant threat from the invasive fungal pathogen <i>Hymenoscyphus fraxineus</i>. Some studies have indicated that the susceptibility of ash to the pathogen is genetically determined, providing some hope for accelerated breeding programs that are aimed at increasing the resistance of ash populations. To address this challenge, we used a genomic selection strategy to identify potential genetic markers that are associated with resistance to the pathogen causing ash dieback. Through genome-wide association studies (GWAS) of 300 common ash individuals from 30 populations across Poland (ddRAD, dataset A), we identified six significant SNP loci with a <i>p</i>-value ≤1 × 10<sup>−4</sup> associated with health status. To further evaluate the effectiveness of GWAS markers in predicting health status, we considered two genomic prediction scenarios. Firstly, we conducted cross-validation on dataset A. Secondly, we trained markers on dataset A and tested them on dataset B, which involved whole-genome sequencing of 20 individuals from two populations. Genomic prediction analysis revealed that the top SNPs identified via GWAS exhibited notably higher prediction accuracies compared to randomly selected SNPs, particularly with a larger number of SNPs. Cross-validation analyses using dataset A showcased high genomic prediction accuracy, predicting tree health status with over 90% accuracy across the top SNP sets ranging from 500 to 10,000 SNPs from the GWAS datasets. However, no significant results emerged for health status when the model trained on dataset A was tested on dataset B. Our findings illuminate potential genetic markers associated with resistance to ash dieback, offering support for future breeding programs in Poland aimed at combating ash dieback and bolstering conservation efforts for this invaluable tree species.</p>\",\"PeriodicalId\":168,\"journal\":{\"name\":\"Evolutionary Applications\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":3.5000,\"publicationDate\":\"2024-05-03\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1111/eva.13694\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Evolutionary Applications\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1111/eva.13694\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"EVOLUTIONARY BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Evolutionary Applications","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/eva.13694","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"EVOLUTIONARY BIOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
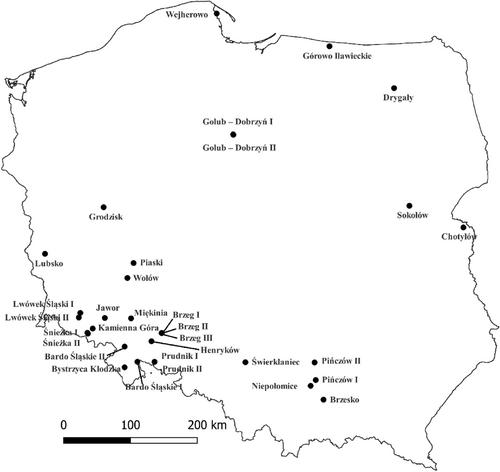
由于人为环境变化而导致的外来虫害和病原体的增加已成为全球树种的一个主要问题。白蜡树(Fraxinus excelsior L.)就是面临入侵真菌病原体Hymenoscyphus fraxineus严重威胁的树种之一。一些研究表明,白蜡对病原体的易感性是由基因决定的,这为旨在提高白蜡种群抗性的加速育种计划带来了一些希望。为了应对这一挑战,我们采用了基因组选择策略,以确定与白蜡树对病原体的抗性相关的潜在遗传标记。通过对来自波兰 30 个种群(ddRAD,数据集 A)的 300 个普通白蜡个体进行全基因组关联研究(GWAS),我们确定了 6 个与健康状况相关的 p 值≤1 × 10-4 的重要 SNP 位点。为了进一步评估 GWAS 标记在预测健康状况方面的有效性,我们考虑了两种基因组预测方案。首先,我们在数据集 A 上进行了交叉验证;其次,我们在数据集 A 上对标记进行了训练,并在数据集 B 上对其进行了测试,数据集 B 涉及来自两个人群的 20 个个体的全基因组测序。基因组预测分析表明,与随机选择的 SNP 相比,通过 GWAS 确定的顶级 SNP 的预测准确率明显更高,尤其是在 SNP 数量较多的情况下。使用数据集 A 进行的交叉验证分析显示了较高的基因组预测准确率,在来自 GWAS 数据集的 500 到 10,000 个 SNP 的顶级 SNP 组中,预测树木健康状况的准确率超过 90%。我们的发现揭示了与白蜡枯萎病抗性相关的潜在遗传标记,为波兰未来旨在防治白蜡枯萎病的育种计划提供了支持,并加强了对这一珍贵树种的保护。
Genomic prediction of resistance to Hymenoscyphus fraxineus in common ash (Fraxinus excelsior L.) populations
The increase in introduced insect pests and pathogens due to anthropogenic environmental changes has become a major concern for tree species worldwide. Common ash (Fraxinus excelsior L.) is one of such species facing a significant threat from the invasive fungal pathogen Hymenoscyphus fraxineus. Some studies have indicated that the susceptibility of ash to the pathogen is genetically determined, providing some hope for accelerated breeding programs that are aimed at increasing the resistance of ash populations. To address this challenge, we used a genomic selection strategy to identify potential genetic markers that are associated with resistance to the pathogen causing ash dieback. Through genome-wide association studies (GWAS) of 300 common ash individuals from 30 populations across Poland (ddRAD, dataset A), we identified six significant SNP loci with a p-value ≤1 × 10−4 associated with health status. To further evaluate the effectiveness of GWAS markers in predicting health status, we considered two genomic prediction scenarios. Firstly, we conducted cross-validation on dataset A. Secondly, we trained markers on dataset A and tested them on dataset B, which involved whole-genome sequencing of 20 individuals from two populations. Genomic prediction analysis revealed that the top SNPs identified via GWAS exhibited notably higher prediction accuracies compared to randomly selected SNPs, particularly with a larger number of SNPs. Cross-validation analyses using dataset A showcased high genomic prediction accuracy, predicting tree health status with over 90% accuracy across the top SNP sets ranging from 500 to 10,000 SNPs from the GWAS datasets. However, no significant results emerged for health status when the model trained on dataset A was tested on dataset B. Our findings illuminate potential genetic markers associated with resistance to ash dieback, offering support for future breeding programs in Poland aimed at combating ash dieback and bolstering conservation efforts for this invaluable tree species.
期刊介绍:
Evolutionary Applications is a fully peer reviewed open access journal. It publishes papers that utilize concepts from evolutionary biology to address biological questions of health, social and economic relevance. Papers are expected to employ evolutionary concepts or methods to make contributions to areas such as (but not limited to): medicine, agriculture, forestry, exploitation and management (fisheries and wildlife), aquaculture, conservation biology, environmental sciences (including climate change and invasion biology), microbiology, and toxicology. All taxonomic groups are covered from microbes, fungi, plants and animals. In order to better serve the community, we also now strongly encourage submissions of papers making use of modern molecular and genetic methods (population and functional genomics, transcriptomics, proteomics, epigenetics, quantitative genetics, association and linkage mapping) to address important questions in any of these disciplines and in an applied evolutionary framework. Theoretical, empirical, synthesis or perspective papers are welcome.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


