Roberta Filograna, Jule Gerlach, Hae-Na Choi, Giovanni Rigoni, Michela Barbaro, Mikael Oscarson, Seungmin Lee, Katarina Tiklova, Markus Ringnér, Camilla Koolmeister, Rolf Wibom, Sara Riggare, Inger Nennesmo, Thomas Perlmann, Anna Wredenberg, Anna Wedell, Elisa Motori, Per Svenningsson, Nils-Göran Larsson
{"title":"成年哺乳动物组织不需要 PARKIN 来维持 OXPHOS 功能","authors":"Roberta Filograna, Jule Gerlach, Hae-Na Choi, Giovanni Rigoni, Michela Barbaro, Mikael Oscarson, Seungmin Lee, Katarina Tiklova, Markus Ringnér, Camilla Koolmeister, Rolf Wibom, Sara Riggare, Inger Nennesmo, Thomas Perlmann, Anna Wredenberg, Anna Wedell, Elisa Motori, Per Svenningsson, Nils-Göran Larsson","doi":"10.1038/s41531-024-00707-0","DOIUrl":null,"url":null,"abstract":"<p>Loss-of-function variants in the <i>PRKN</i> gene encoding the ubiquitin E3 ligase PARKIN cause autosomal recessive early-onset Parkinson’s disease (PD). Extensive in vitro and in vivo studies have reported that PARKIN is involved in multiple pathways of mitochondrial quality control, including mitochondrial degradation and biogenesis. However, these findings are surrounded by substantial controversy due to conflicting experimental data. In addition, the existing PARKIN-deficient mouse models have failed to faithfully recapitulate PD phenotypes. Therefore, we have investigated the mitochondrial role of PARKIN during ageing and in response to stress by employing a series of conditional Parkin knockout mice. We report that PARKIN loss does not affect oxidative phosphorylation (OXPHOS) capacity and mitochondrial DNA (mtDNA) levels in the brain, heart, and skeletal muscle of aged mice. We also demonstrate that PARKIN deficiency does not exacerbate the brain defects and the pro-inflammatory phenotype observed in mice carrying high levels of mtDNA mutations. To rule out compensatory mechanisms activated during embryonic development of <i>Parkin</i>-deficient mice, we generated a mouse model where loss of PARKIN was induced in adult dopaminergic (DA) neurons. Surprisingly, also these mice did not show motor impairment or neurodegeneration, and no major transcriptional changes were found in isolated midbrain DA neurons. Finally, we report a patient with compound heterozygous <i>PRKN</i> pathogenic variants that lacks PARKIN and has developed PD. The PARKIN deficiency did not impair OXPHOS activities or induce mitochondrial pathology in skeletal muscle from the patient. Altogether, our results argue that PARKIN is dispensable for OXPHOS function in adult mammalian tissues.</p>","PeriodicalId":19706,"journal":{"name":"NPJ Parkinson's Disease","volume":null,"pages":null},"PeriodicalIF":6.7000,"publicationDate":"2024-04-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"PARKIN is not required to sustain OXPHOS function in adult mammalian tissues\",\"authors\":\"Roberta Filograna, Jule Gerlach, Hae-Na Choi, Giovanni Rigoni, Michela Barbaro, Mikael Oscarson, Seungmin Lee, Katarina Tiklova, Markus Ringnér, Camilla Koolmeister, Rolf Wibom, Sara Riggare, Inger Nennesmo, Thomas Perlmann, Anna Wredenberg, Anna Wedell, Elisa Motori, Per Svenningsson, Nils-Göran Larsson\",\"doi\":\"10.1038/s41531-024-00707-0\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Loss-of-function variants in the <i>PRKN</i> gene encoding the ubiquitin E3 ligase PARKIN cause autosomal recessive early-onset Parkinson’s disease (PD). Extensive in vitro and in vivo studies have reported that PARKIN is involved in multiple pathways of mitochondrial quality control, including mitochondrial degradation and biogenesis. However, these findings are surrounded by substantial controversy due to conflicting experimental data. In addition, the existing PARKIN-deficient mouse models have failed to faithfully recapitulate PD phenotypes. Therefore, we have investigated the mitochondrial role of PARKIN during ageing and in response to stress by employing a series of conditional Parkin knockout mice. We report that PARKIN loss does not affect oxidative phosphorylation (OXPHOS) capacity and mitochondrial DNA (mtDNA) levels in the brain, heart, and skeletal muscle of aged mice. We also demonstrate that PARKIN deficiency does not exacerbate the brain defects and the pro-inflammatory phenotype observed in mice carrying high levels of mtDNA mutations. To rule out compensatory mechanisms activated during embryonic development of <i>Parkin</i>-deficient mice, we generated a mouse model where loss of PARKIN was induced in adult dopaminergic (DA) neurons. Surprisingly, also these mice did not show motor impairment or neurodegeneration, and no major transcriptional changes were found in isolated midbrain DA neurons. Finally, we report a patient with compound heterozygous <i>PRKN</i> pathogenic variants that lacks PARKIN and has developed PD. The PARKIN deficiency did not impair OXPHOS activities or induce mitochondrial pathology in skeletal muscle from the patient. Altogether, our results argue that PARKIN is dispensable for OXPHOS function in adult mammalian tissues.</p>\",\"PeriodicalId\":19706,\"journal\":{\"name\":\"NPJ Parkinson's Disease\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":6.7000,\"publicationDate\":\"2024-04-29\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"NPJ Parkinson's Disease\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1038/s41531-024-00707-0\",\"RegionNum\":1,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"NEUROSCIENCES\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"NPJ Parkinson's Disease","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1038/s41531-024-00707-0","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"NEUROSCIENCES","Score":null,"Total":0}
PARKIN is not required to sustain OXPHOS function in adult mammalian tissues
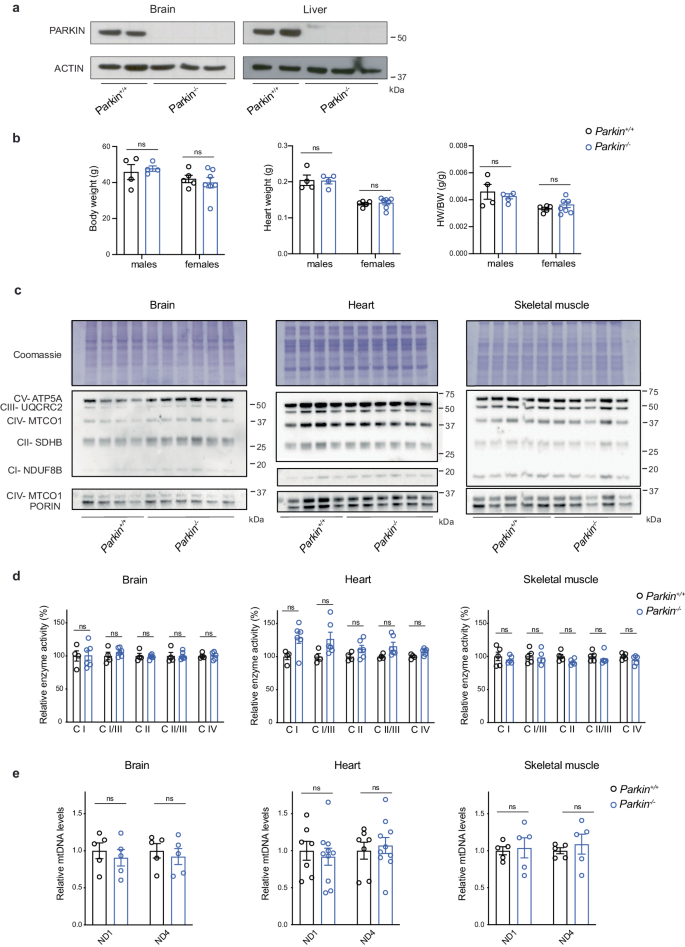
Loss-of-function variants in the PRKN gene encoding the ubiquitin E3 ligase PARKIN cause autosomal recessive early-onset Parkinson’s disease (PD). Extensive in vitro and in vivo studies have reported that PARKIN is involved in multiple pathways of mitochondrial quality control, including mitochondrial degradation and biogenesis. However, these findings are surrounded by substantial controversy due to conflicting experimental data. In addition, the existing PARKIN-deficient mouse models have failed to faithfully recapitulate PD phenotypes. Therefore, we have investigated the mitochondrial role of PARKIN during ageing and in response to stress by employing a series of conditional Parkin knockout mice. We report that PARKIN loss does not affect oxidative phosphorylation (OXPHOS) capacity and mitochondrial DNA (mtDNA) levels in the brain, heart, and skeletal muscle of aged mice. We also demonstrate that PARKIN deficiency does not exacerbate the brain defects and the pro-inflammatory phenotype observed in mice carrying high levels of mtDNA mutations. To rule out compensatory mechanisms activated during embryonic development of Parkin-deficient mice, we generated a mouse model where loss of PARKIN was induced in adult dopaminergic (DA) neurons. Surprisingly, also these mice did not show motor impairment or neurodegeneration, and no major transcriptional changes were found in isolated midbrain DA neurons. Finally, we report a patient with compound heterozygous PRKN pathogenic variants that lacks PARKIN and has developed PD. The PARKIN deficiency did not impair OXPHOS activities or induce mitochondrial pathology in skeletal muscle from the patient. Altogether, our results argue that PARKIN is dispensable for OXPHOS function in adult mammalian tissues.
期刊介绍:
npj Parkinson's Disease is a comprehensive open access journal that covers a wide range of research areas related to Parkinson's disease. It publishes original studies in basic science, translational research, and clinical investigations. The journal is dedicated to advancing our understanding of Parkinson's disease by exploring various aspects such as anatomy, etiology, genetics, cellular and molecular physiology, neurophysiology, epidemiology, and therapeutic development. By providing free and immediate access to the scientific and Parkinson's disease community, npj Parkinson's Disease promotes collaboration and knowledge sharing among researchers and healthcare professionals.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


