Claire Freycon, Edith Sepulchre, Vincent-Philippe Lavallée, David Mitchell, Margaret L. MacMillan, Catherine Vezina, Catherine Goudie
{"title":"小儿急性早幼粒细胞白血病和范可尼贫血:病例报告和文献综述。","authors":"Claire Freycon, Edith Sepulchre, Vincent-Philippe Lavallée, David Mitchell, Margaret L. MacMillan, Catherine Vezina, Catherine Goudie","doi":"10.1111/cge.14537","DOIUrl":null,"url":null,"abstract":"<p>Acute promyelocytic leukemia (APL) represents 5%–10% of childhood acute myeloid leukemia (AML) and is the most curable subtype of AML. Fanconi anemia (FA) is one of the most common inherited bone marrow failure syndromes caused by biallelic pathogenic variants (PV) in specific DNA-repair genes. Biallelic PVs in <i>FANCD1/BRCA2</i> (FA-D1) account for 3% of FA and are associated with early-onset leukemia and a high risk of solid tumors. We report a 4 year-old boy from non-consanguineous parents diagnosed with standard risk APL. This child had café-au-lait spots and an extra thumb remnant. Genomic sequencing revealed two PV in <i>FANCD1/BRCA2</i> confirming a diagnosis of FA-D1. Chromosomal breakage studies were compatible with FA. Each parent carried one variant and had no personal history of cancer. Morphological then molecular remissions were achieved with all-trans retinoic acid and Arsenic trioxide. This patient underwent haploidentical stem cell transplant. In addition to our patient, a literature search revealed four additional patients with APL/FA, with a total of three patients with FA-D1. This raises the possibility of an association between such rare disorders. Practical management of APL in the setting of FA-D1 is discussed with an overview of current evidence and knowledge gaps.</p>","PeriodicalId":10354,"journal":{"name":"Clinical Genetics","volume":null,"pages":null},"PeriodicalIF":2.9000,"publicationDate":"2024-04-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Pediatric acute promyelocytic leukemia and Fanconi anemia: Case report and literature review\",\"authors\":\"Claire Freycon, Edith Sepulchre, Vincent-Philippe Lavallée, David Mitchell, Margaret L. MacMillan, Catherine Vezina, Catherine Goudie\",\"doi\":\"10.1111/cge.14537\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Acute promyelocytic leukemia (APL) represents 5%–10% of childhood acute myeloid leukemia (AML) and is the most curable subtype of AML. Fanconi anemia (FA) is one of the most common inherited bone marrow failure syndromes caused by biallelic pathogenic variants (PV) in specific DNA-repair genes. Biallelic PVs in <i>FANCD1/BRCA2</i> (FA-D1) account for 3% of FA and are associated with early-onset leukemia and a high risk of solid tumors. We report a 4 year-old boy from non-consanguineous parents diagnosed with standard risk APL. This child had café-au-lait spots and an extra thumb remnant. Genomic sequencing revealed two PV in <i>FANCD1/BRCA2</i> confirming a diagnosis of FA-D1. Chromosomal breakage studies were compatible with FA. Each parent carried one variant and had no personal history of cancer. Morphological then molecular remissions were achieved with all-trans retinoic acid and Arsenic trioxide. This patient underwent haploidentical stem cell transplant. In addition to our patient, a literature search revealed four additional patients with APL/FA, with a total of three patients with FA-D1. This raises the possibility of an association between such rare disorders. Practical management of APL in the setting of FA-D1 is discussed with an overview of current evidence and knowledge gaps.</p>\",\"PeriodicalId\":10354,\"journal\":{\"name\":\"Clinical Genetics\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2024-04-24\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Clinical Genetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1111/cge.14537\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Genetics","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/cge.14537","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
摘要
急性早幼粒细胞白血病(APL)占儿童急性髓细胞白血病(AML)的5%-10%,是AML中最容易治愈的亚型。范可尼贫血(Fanconi anemia,FA)是最常见的遗传性骨髓衰竭综合征之一,由特定 DNA 修复基因中的双叶致病变体(PV)引起。FANCD1/BRCA2(FA-D1)中的双拷贝致病变体占 FA 的 3%,与早发型白血病和实体瘤的高风险有关。我们报告了一名非近亲结婚的 4 岁男孩,他被诊断为标准风险 APL。该患儿有咖啡斑和一个额外的拇指残余。基因组测序发现 FANCD1/BRCA2 中有两个 PV,确诊为 FA-D1。染色体断裂研究与 FA 相吻合。父母双方各携带一个变异基因,没有个人癌症病史。使用全反式维甲酸和三氧化二砷后,患者的形态和分子症状均得到缓解。这名患者接受了单倍体干细胞移植。除我们的患者外,文献检索还发现了另外四名 APL/FA 患者,其中共有三名 FA-D1 患者。这就提出了这种罕见疾病之间存在关联的可能性。本文讨论了在FA-D1情况下对APL的实际管理,并概述了目前的证据和知识差距。
Pediatric acute promyelocytic leukemia and Fanconi anemia: Case report and literature review
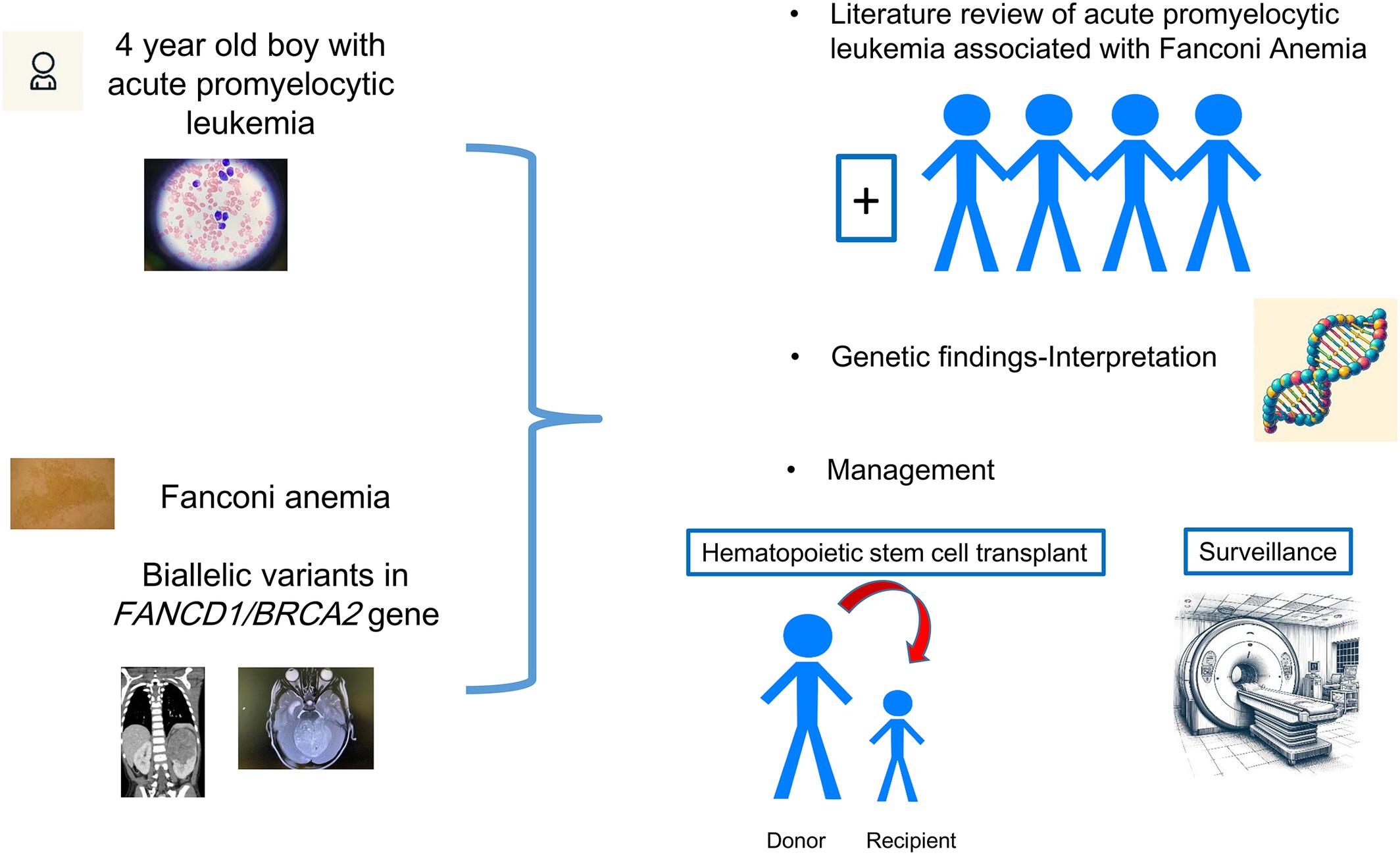
Acute promyelocytic leukemia (APL) represents 5%–10% of childhood acute myeloid leukemia (AML) and is the most curable subtype of AML. Fanconi anemia (FA) is one of the most common inherited bone marrow failure syndromes caused by biallelic pathogenic variants (PV) in specific DNA-repair genes. Biallelic PVs in FANCD1/BRCA2 (FA-D1) account for 3% of FA and are associated with early-onset leukemia and a high risk of solid tumors. We report a 4 year-old boy from non-consanguineous parents diagnosed with standard risk APL. This child had café-au-lait spots and an extra thumb remnant. Genomic sequencing revealed two PV in FANCD1/BRCA2 confirming a diagnosis of FA-D1. Chromosomal breakage studies were compatible with FA. Each parent carried one variant and had no personal history of cancer. Morphological then molecular remissions were achieved with all-trans retinoic acid and Arsenic trioxide. This patient underwent haploidentical stem cell transplant. In addition to our patient, a literature search revealed four additional patients with APL/FA, with a total of three patients with FA-D1. This raises the possibility of an association between such rare disorders. Practical management of APL in the setting of FA-D1 is discussed with an overview of current evidence and knowledge gaps.
期刊介绍:
Clinical Genetics links research to the clinic, translating advances in our understanding of the molecular basis of genetic disease for the practising clinical geneticist. The journal publishes high quality research papers, short reports, reviews and mini-reviews that connect medical genetics research with clinical practice.
Topics of particular interest are:
• Linking genetic variations to disease
• Genome rearrangements and disease
• Epigenetics and disease
• The translation of genotype to phenotype
• Genetics of complex disease
• Management/intervention of genetic diseases
• Novel therapies for genetic diseases
• Developmental biology, as it relates to clinical genetics
• Social science research on the psychological and behavioural aspects of living with or being at risk of genetic disease

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


