{"title":"用自适应核电子轨道方法优化量子核位置","authors":"Lukas Hasecke*, and , Ricardo A. Mata*, ","doi":"10.1021/acs.jpca.4c00096","DOIUrl":null,"url":null,"abstract":"<p >The use of multicomponent methods has become increasingly popular over the last years. Under this framework, nuclei (commonly protons) are treated quantum mechanically on the same footing as the electronic structure problem. Under the use of atomic-centered orbitals, this can lead to some complications as the ideal location of the nuclear basis centers must be optimized. In this contribution, we propose a straightforward approach to determine the position of such centers within the self-consistent cycle of a multicomponent calculation, making use of individual proton charge centroids. We test the method on model systems including the water dimer, a protonated water tetramer, and a porphine system. Comparing to numerical gradient calculations, the adaptive nuclear-electronic orbital (NEO) procedure is able to converge the basis centers to within a few cents of an Ångström and with less than 0.1 kcal/mol differences in absolute energies. This is achieved in one single calculation and with a small added computational effort of up to 80% compared to a regular NEO- self-consistent field run. An example application for the human transketolase proton wire is also provided.</p>","PeriodicalId":59,"journal":{"name":"The Journal of Physical Chemistry A","volume":"128 16","pages":"3205–3211"},"PeriodicalIF":2.8000,"publicationDate":"2024-04-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.acs.org/doi/epdf/10.1021/acs.jpca.4c00096","citationCount":"0","resultStr":"{\"title\":\"Optimization of Quantum Nuclei Positions with the Adaptive Nuclear-Electronic Orbital Approach\",\"authors\":\"Lukas Hasecke*, and , Ricardo A. Mata*, \",\"doi\":\"10.1021/acs.jpca.4c00096\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >The use of multicomponent methods has become increasingly popular over the last years. Under this framework, nuclei (commonly protons) are treated quantum mechanically on the same footing as the electronic structure problem. Under the use of atomic-centered orbitals, this can lead to some complications as the ideal location of the nuclear basis centers must be optimized. In this contribution, we propose a straightforward approach to determine the position of such centers within the self-consistent cycle of a multicomponent calculation, making use of individual proton charge centroids. We test the method on model systems including the water dimer, a protonated water tetramer, and a porphine system. Comparing to numerical gradient calculations, the adaptive nuclear-electronic orbital (NEO) procedure is able to converge the basis centers to within a few cents of an Ångström and with less than 0.1 kcal/mol differences in absolute energies. This is achieved in one single calculation and with a small added computational effort of up to 80% compared to a regular NEO- self-consistent field run. An example application for the human transketolase proton wire is also provided.</p>\",\"PeriodicalId\":59,\"journal\":{\"name\":\"The Journal of Physical Chemistry A\",\"volume\":\"128 16\",\"pages\":\"3205–3211\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2024-04-15\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.acs.org/doi/epdf/10.1021/acs.jpca.4c00096\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry A\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jpca.4c00096\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry A","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpca.4c00096","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Optimization of Quantum Nuclei Positions with the Adaptive Nuclear-Electronic Orbital Approach
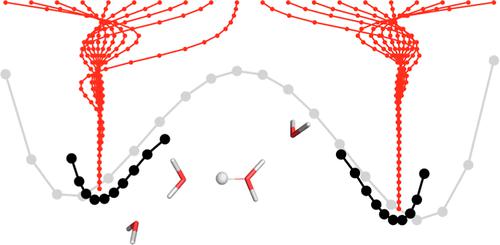
The use of multicomponent methods has become increasingly popular over the last years. Under this framework, nuclei (commonly protons) are treated quantum mechanically on the same footing as the electronic structure problem. Under the use of atomic-centered orbitals, this can lead to some complications as the ideal location of the nuclear basis centers must be optimized. In this contribution, we propose a straightforward approach to determine the position of such centers within the self-consistent cycle of a multicomponent calculation, making use of individual proton charge centroids. We test the method on model systems including the water dimer, a protonated water tetramer, and a porphine system. Comparing to numerical gradient calculations, the adaptive nuclear-electronic orbital (NEO) procedure is able to converge the basis centers to within a few cents of an Ångström and with less than 0.1 kcal/mol differences in absolute energies. This is achieved in one single calculation and with a small added computational effort of up to 80% compared to a regular NEO- self-consistent field run. An example application for the human transketolase proton wire is also provided.
期刊介绍:
The Journal of Physical Chemistry A is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


