Nan Xu, Petter Rosander, Christian Schäfer, Eric Lindgren, Nicklas Österbacka, Mandi Fang, Wei Chen, Yi He*, Zheyong Fan* and Paul Erhart*,
{"title":"通过神经进化势框架获得张量特性:红外和拉曼光谱的快速模拟","authors":"Nan Xu, Petter Rosander, Christian Schäfer, Eric Lindgren, Nicklas Österbacka, Mandi Fang, Wei Chen, Yi He*, Zheyong Fan* and Paul Erhart*, ","doi":"10.1021/acs.jctc.3c01343","DOIUrl":null,"url":null,"abstract":"<p >Infrared and Raman spectroscopy are widely used for the characterization of gases, liquids, and solids, as the spectra contain a wealth of information concerning, in particular, the dynamics of these systems. Atomic scale simulations can be used to predict such spectra but are often severely limited due to high computational cost or the need for strong approximations that limit the application range and reliability. Here, we introduce a machine learning (ML) accelerated approach that addresses these shortcomings and provides a significant performance boost in terms of data and computational efficiency compared with earlier ML schemes. To this end, we generalize the neuroevolution potential approach to enable the prediction of rank one and two tensors to obtain the tensorial neuroevolution potential (TNEP) scheme. We apply the resulting framework to construct models for the dipole moment, polarizability, and susceptibility of molecules, liquids, and solids and show that our approach compares favorably with several ML models from the literature with respect to accuracy and computational efficiency. Finally, we demonstrate the application of the TNEP approach to the prediction of infrared and Raman spectra of liquid water, a molecule (PTAF<sup>–</sup>), and a prototypical perovskite with strong anharmonicity (BaZrO<sub>3</sub>). The TNEP approach is implemented in the free and open source software package <span>gpumd</span>, which makes this methodology readily available to the scientific community.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"20 8","pages":"3273–3284"},"PeriodicalIF":5.5000,"publicationDate":"2024-04-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.acs.org/doi/epdf/10.1021/acs.jctc.3c01343","citationCount":"0","resultStr":"{\"title\":\"Tensorial Properties via the Neuroevolution Potential Framework: Fast Simulation of Infrared and Raman Spectra\",\"authors\":\"Nan Xu, Petter Rosander, Christian Schäfer, Eric Lindgren, Nicklas Österbacka, Mandi Fang, Wei Chen, Yi He*, Zheyong Fan* and Paul Erhart*, \",\"doi\":\"10.1021/acs.jctc.3c01343\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Infrared and Raman spectroscopy are widely used for the characterization of gases, liquids, and solids, as the spectra contain a wealth of information concerning, in particular, the dynamics of these systems. Atomic scale simulations can be used to predict such spectra but are often severely limited due to high computational cost or the need for strong approximations that limit the application range and reliability. Here, we introduce a machine learning (ML) accelerated approach that addresses these shortcomings and provides a significant performance boost in terms of data and computational efficiency compared with earlier ML schemes. To this end, we generalize the neuroevolution potential approach to enable the prediction of rank one and two tensors to obtain the tensorial neuroevolution potential (TNEP) scheme. We apply the resulting framework to construct models for the dipole moment, polarizability, and susceptibility of molecules, liquids, and solids and show that our approach compares favorably with several ML models from the literature with respect to accuracy and computational efficiency. Finally, we demonstrate the application of the TNEP approach to the prediction of infrared and Raman spectra of liquid water, a molecule (PTAF<sup>–</sup>), and a prototypical perovskite with strong anharmonicity (BaZrO<sub>3</sub>). The TNEP approach is implemented in the free and open source software package <span>gpumd</span>, which makes this methodology readily available to the scientific community.</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\"20 8\",\"pages\":\"3273–3284\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2024-04-04\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.acs.org/doi/epdf/10.1021/acs.jctc.3c01343\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jctc.3c01343\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.3c01343","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
摘要
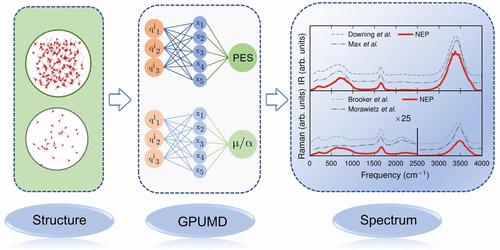
红外光谱和拉曼光谱被广泛应用于气体、液体和固体的表征,因为光谱包含大量信息,特别是这些系统的动力学信息。原子尺度模拟可用于预测此类光谱,但由于计算成本高昂或需要进行强近似,其应用范围和可靠性往往受到严重限制。在此,我们介绍一种机器学习(ML)加速方法,它能解决这些不足之处,与早期的 ML 方案相比,在数据和计算效率方面有显著的性能提升。为此,我们推广了神经进化势方法,使其能够预测一阶和二阶张量,从而获得了张量神经进化势(TNEP)方案。我们将由此产生的框架用于构建分子、液体和固体的偶极矩、极化性和电感模型,并证明我们的方法在准确性和计算效率方面优于文献中的几种 ML 模型。最后,我们展示了 TNEP 方法在预测液态水、分子(PTAF-)和具有强非谐波性的原型包晶(BaZrO3)的红外和拉曼光谱中的应用。TNEP 方法是在免费开源软件包 gpumd 中实现的,因此科学界可以随时使用这种方法。
Tensorial Properties via the Neuroevolution Potential Framework: Fast Simulation of Infrared and Raman Spectra
Infrared and Raman spectroscopy are widely used for the characterization of gases, liquids, and solids, as the spectra contain a wealth of information concerning, in particular, the dynamics of these systems. Atomic scale simulations can be used to predict such spectra but are often severely limited due to high computational cost or the need for strong approximations that limit the application range and reliability. Here, we introduce a machine learning (ML) accelerated approach that addresses these shortcomings and provides a significant performance boost in terms of data and computational efficiency compared with earlier ML schemes. To this end, we generalize the neuroevolution potential approach to enable the prediction of rank one and two tensors to obtain the tensorial neuroevolution potential (TNEP) scheme. We apply the resulting framework to construct models for the dipole moment, polarizability, and susceptibility of molecules, liquids, and solids and show that our approach compares favorably with several ML models from the literature with respect to accuracy and computational efficiency. Finally, we demonstrate the application of the TNEP approach to the prediction of infrared and Raman spectra of liquid water, a molecule (PTAF–), and a prototypical perovskite with strong anharmonicity (BaZrO3). The TNEP approach is implemented in the free and open source software package gpumd, which makes this methodology readily available to the scientific community.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


