Kevin D Wolter, Asayuki Kamatani, Yumiko Suzuki, Takayuki Imaeda, Ramzi Dagher, Allan Safferman, Rod Junor
{"title":"从赞助商的角度看监管机构要求的上市后观察性研究对了解日本人体用药产品效益/风险的贡献。","authors":"Kevin D Wolter, Asayuki Kamatani, Yumiko Suzuki, Takayuki Imaeda, Ramzi Dagher, Allan Safferman, Rod Junor","doi":"10.1007/s40290-024-00521-2","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Following marketing authorization in Japan, for almost all new drugs or new indications, postmarketing studies (PMS) are a regulatory requirement. These PMS focus on accrual of a defined number of cases with data being collected for a predetermined period after approval to confirm efficacy/effectiveness, safety, and quality in the Japanese population. In contrast to other regions where PMS are only required to address a specific scientific uncertainty, in Japan, PMS are often required regardless of any specific scientific uncertainty, and therefore, their scientific value is unclear.</p><p><strong>Objectives: </strong>To determine the contribution to the understanding of benefit/risk of PMS conducted by Pfizer in Japan over 2000-2020 for Pharmaceuticals and Medical Devices Agency (PMDA) reexamination.</p><p><strong>Methods: </strong>A retrospective analysis of all Pfizer Japan postmarketing studies (PMS) during 2000-2020 was performed. Available Pfizer clinical study reports (CSRs) and PMDA reexamination reports (RERs) were reviewed for key safety findings. The primary analysis was conducted on the subset of PMS that had both an English CSR and a discussion of that PMS in the relevant RER issued by the PMDA, which was subsequently translated into English by a professional translation vendor. Reexamination outcome is included in each RER and served to demonstrate the impact of the study of the benefit/risk profile of the drug.</p><p><strong>Results: </strong>A total of 79 PMS for 43 different drug products across therapy areas enrolled a total of 98,035 patients. The 79 PMS comprised 34 general drug use investigation (GDUI) studies and 45 special investigation (SI) studies. The primary analysis involved 37 PMS with a CSR and RER available in English (40,470 patients); all of which were observational in design. For 31 of 37 PMS, the RER concluded the overall adverse drug reaction (ADR) rate in the PMS was nominally lower than in the phase 3 program. Unlabeled ADRs were reported in 28 of 37 PMS; however, no new safety concerns requiring regulatory action arose from any PMS. The PMDA did not require additional risk minimization measures for any of the 43 drug products studied in any of the 79 PMS assessed. Japan PMS data were consistent with prior global data with no evidence of clinically meaningful differences in safety in Japanese patients. In all cases, the reexamination outcome was category 1 (\"usefulness is confirmed\").</p><p><strong>Conclusions: </strong>The reexamination process did not result in regulatory changes for any of the examined drugs. The Japan new-drug application (J-NDA) review and approval process, including implementation of the initial Japan product label, assures acceptable benefit/risk at the time of approval such that mandatory GDUI or SI studies for all products should be reconsidered. In the case of genuine scientific uncertainty to the extent that the benefit/risk of the product is not clear, a PMS is warranted.</p>","PeriodicalId":19778,"journal":{"name":"Pharmaceutical Medicine","volume":" ","pages":"217-224"},"PeriodicalIF":4.5000,"publicationDate":"2024-05-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11101524/pdf/","citationCount":"0","resultStr":"{\"title\":\"A Sponsor's Perspective on the Contribution of Regulatory-Required Observational Post-Marketing Studies to Understanding Human Drug Product Benefit/Risk in Japan.\",\"authors\":\"Kevin D Wolter, Asayuki Kamatani, Yumiko Suzuki, Takayuki Imaeda, Ramzi Dagher, Allan Safferman, Rod Junor\",\"doi\":\"10.1007/s40290-024-00521-2\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Following marketing authorization in Japan, for almost all new drugs or new indications, postmarketing studies (PMS) are a regulatory requirement. These PMS focus on accrual of a defined number of cases with data being collected for a predetermined period after approval to confirm efficacy/effectiveness, safety, and quality in the Japanese population. In contrast to other regions where PMS are only required to address a specific scientific uncertainty, in Japan, PMS are often required regardless of any specific scientific uncertainty, and therefore, their scientific value is unclear.</p><p><strong>Objectives: </strong>To determine the contribution to the understanding of benefit/risk of PMS conducted by Pfizer in Japan over 2000-2020 for Pharmaceuticals and Medical Devices Agency (PMDA) reexamination.</p><p><strong>Methods: </strong>A retrospective analysis of all Pfizer Japan postmarketing studies (PMS) during 2000-2020 was performed. Available Pfizer clinical study reports (CSRs) and PMDA reexamination reports (RERs) were reviewed for key safety findings. The primary analysis was conducted on the subset of PMS that had both an English CSR and a discussion of that PMS in the relevant RER issued by the PMDA, which was subsequently translated into English by a professional translation vendor. Reexamination outcome is included in each RER and served to demonstrate the impact of the study of the benefit/risk profile of the drug.</p><p><strong>Results: </strong>A total of 79 PMS for 43 different drug products across therapy areas enrolled a total of 98,035 patients. The 79 PMS comprised 34 general drug use investigation (GDUI) studies and 45 special investigation (SI) studies. The primary analysis involved 37 PMS with a CSR and RER available in English (40,470 patients); all of which were observational in design. For 31 of 37 PMS, the RER concluded the overall adverse drug reaction (ADR) rate in the PMS was nominally lower than in the phase 3 program. Unlabeled ADRs were reported in 28 of 37 PMS; however, no new safety concerns requiring regulatory action arose from any PMS. The PMDA did not require additional risk minimization measures for any of the 43 drug products studied in any of the 79 PMS assessed. Japan PMS data were consistent with prior global data with no evidence of clinically meaningful differences in safety in Japanese patients. In all cases, the reexamination outcome was category 1 (\\\"usefulness is confirmed\\\").</p><p><strong>Conclusions: </strong>The reexamination process did not result in regulatory changes for any of the examined drugs. The Japan new-drug application (J-NDA) review and approval process, including implementation of the initial Japan product label, assures acceptable benefit/risk at the time of approval such that mandatory GDUI or SI studies for all products should be reconsidered. In the case of genuine scientific uncertainty to the extent that the benefit/risk of the product is not clear, a PMS is warranted.</p>\",\"PeriodicalId\":19778,\"journal\":{\"name\":\"Pharmaceutical Medicine\",\"volume\":\" \",\"pages\":\"217-224\"},\"PeriodicalIF\":4.5000,\"publicationDate\":\"2024-05-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11101524/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Pharmaceutical Medicine\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1007/s40290-024-00521-2\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/3/31 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"PHARMACOLOGY & PHARMACY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Pharmaceutical Medicine","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1007/s40290-024-00521-2","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/3/31 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"PHARMACOLOGY & PHARMACY","Score":null,"Total":0}
A Sponsor's Perspective on the Contribution of Regulatory-Required Observational Post-Marketing Studies to Understanding Human Drug Product Benefit/Risk in Japan.
Background: Following marketing authorization in Japan, for almost all new drugs or new indications, postmarketing studies (PMS) are a regulatory requirement. These PMS focus on accrual of a defined number of cases with data being collected for a predetermined period after approval to confirm efficacy/effectiveness, safety, and quality in the Japanese population. In contrast to other regions where PMS are only required to address a specific scientific uncertainty, in Japan, PMS are often required regardless of any specific scientific uncertainty, and therefore, their scientific value is unclear.
Objectives: To determine the contribution to the understanding of benefit/risk of PMS conducted by Pfizer in Japan over 2000-2020 for Pharmaceuticals and Medical Devices Agency (PMDA) reexamination.
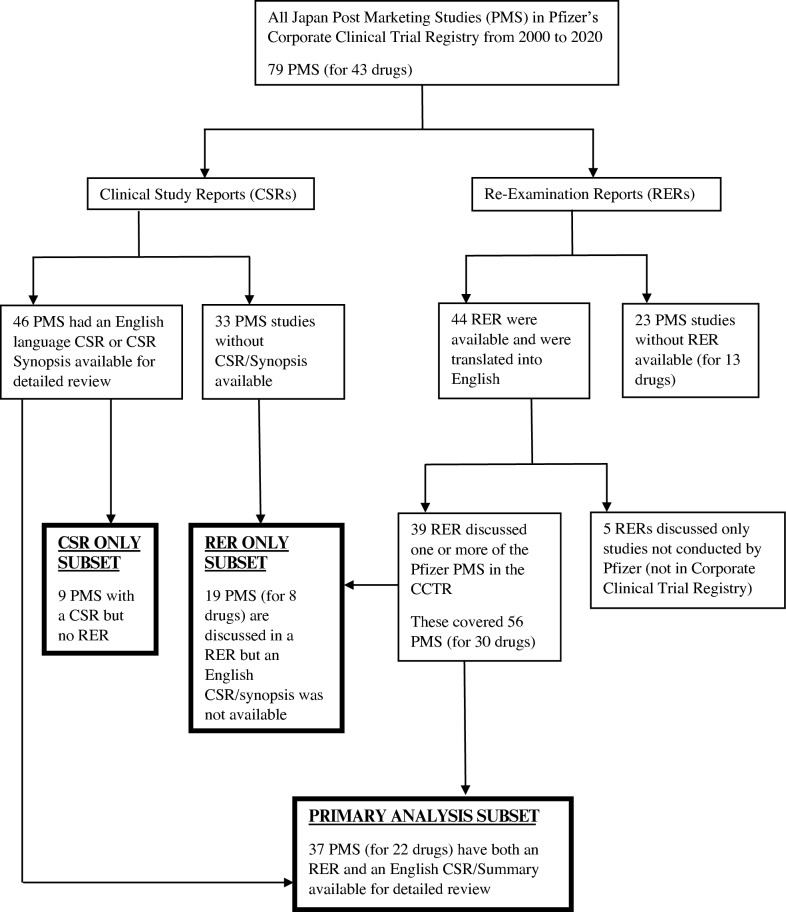
Methods: A retrospective analysis of all Pfizer Japan postmarketing studies (PMS) during 2000-2020 was performed. Available Pfizer clinical study reports (CSRs) and PMDA reexamination reports (RERs) were reviewed for key safety findings. The primary analysis was conducted on the subset of PMS that had both an English CSR and a discussion of that PMS in the relevant RER issued by the PMDA, which was subsequently translated into English by a professional translation vendor. Reexamination outcome is included in each RER and served to demonstrate the impact of the study of the benefit/risk profile of the drug.
Results: A total of 79 PMS for 43 different drug products across therapy areas enrolled a total of 98,035 patients. The 79 PMS comprised 34 general drug use investigation (GDUI) studies and 45 special investigation (SI) studies. The primary analysis involved 37 PMS with a CSR and RER available in English (40,470 patients); all of which were observational in design. For 31 of 37 PMS, the RER concluded the overall adverse drug reaction (ADR) rate in the PMS was nominally lower than in the phase 3 program. Unlabeled ADRs were reported in 28 of 37 PMS; however, no new safety concerns requiring regulatory action arose from any PMS. The PMDA did not require additional risk minimization measures for any of the 43 drug products studied in any of the 79 PMS assessed. Japan PMS data were consistent with prior global data with no evidence of clinically meaningful differences in safety in Japanese patients. In all cases, the reexamination outcome was category 1 ("usefulness is confirmed").
Conclusions: The reexamination process did not result in regulatory changes for any of the examined drugs. The Japan new-drug application (J-NDA) review and approval process, including implementation of the initial Japan product label, assures acceptable benefit/risk at the time of approval such that mandatory GDUI or SI studies for all products should be reconsidered. In the case of genuine scientific uncertainty to the extent that the benefit/risk of the product is not clear, a PMS is warranted.
期刊介绍:
Pharmaceutical Medicine is a specialist discipline concerned with medical aspects of the discovery, development, evaluation, registration, regulation, monitoring, marketing, distribution and pricing of medicines, drug-device and drug-diagnostic combinations. The Journal disseminates information to support the community of professionals working in these highly inter-related functions. Key areas include translational medicine, clinical trial design, pharmacovigilance, clinical toxicology, drug regulation, clinical pharmacology, biostatistics and pharmacoeconomics. The Journal includes:Overviews of contentious or emerging issues.Comprehensive narrative reviews that provide an authoritative source of information on topical issues.Systematic reviews that collate empirical evidence to answer a specific research question, using explicit, systematic methods as outlined by PRISMA statement.Original research articles reporting the results of well-designed studies with a strong link to wider areas of clinical research.Additional digital features (including animated abstracts, video abstracts, slide decks, audio slides, instructional videos, infographics, podcasts and animations) can be published with articles; these are designed to increase the visibility, readership and educational value of the journal’s content. In addition, articles published in Pharmaceutical Medicine may be accompanied by plain language summaries to assist readers who have some knowledge of, but not in-depth expertise in, the area to understand important medical advances.All manuscripts are subject to peer review by international experts. Letters to the Editor are welcomed and will be considered for publication.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


