{"title":"使用 Ab Initio、基于 DFT 和半经验方法模拟 C60 富勒烯中的高次谐波发生。","authors":"Aleksander P. Woźniak*, and , Robert Moszyński, ","doi":"10.1021/acs.jpca.3c07865","DOIUrl":null,"url":null,"abstract":"<p >We report calculations of the high-harmonic generation spectra of the C<sub>60</sub> fullerene molecule carried out by employing a diverse set of real-time time-dependent quantum chemical methods. All methodologies involve expanding the propagated electronic wave function in bases consisting of the ground and singly excited time-independent eigenstates obtained through the solution of the corresponding linear-response equations. We identify the correlation and exchange effect in the spectra by comparing the results from methods relying on the Hartree–Fock reference determinant with those obtained using approaches based on the density functional theory with different exchange–correlation functionals. The effect of the full random-phase approximation treatment of the excited electronic states is also analyzed and compared with the configuration interaction singles and the Tamm–Dancoff approximation. We also showcase the fact that the real-time extension of the semiempirical method INDO/S can be effectively applied for an approximate description of laser-driven dynamics in large systems.</p>","PeriodicalId":59,"journal":{"name":"The Journal of Physical Chemistry A","volume":"128 14","pages":"2683–2702"},"PeriodicalIF":2.8000,"publicationDate":"2024-03-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.acs.org/doi/epdf/10.1021/acs.jpca.3c07865","citationCount":"0","resultStr":"{\"title\":\"Modeling of High-Harmonic Generation in the C60 Fullerene Using Ab Initio, DFT-Based, and Semiempirical Methods\",\"authors\":\"Aleksander P. Woźniak*, and , Robert Moszyński, \",\"doi\":\"10.1021/acs.jpca.3c07865\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >We report calculations of the high-harmonic generation spectra of the C<sub>60</sub> fullerene molecule carried out by employing a diverse set of real-time time-dependent quantum chemical methods. All methodologies involve expanding the propagated electronic wave function in bases consisting of the ground and singly excited time-independent eigenstates obtained through the solution of the corresponding linear-response equations. We identify the correlation and exchange effect in the spectra by comparing the results from methods relying on the Hartree–Fock reference determinant with those obtained using approaches based on the density functional theory with different exchange–correlation functionals. The effect of the full random-phase approximation treatment of the excited electronic states is also analyzed and compared with the configuration interaction singles and the Tamm–Dancoff approximation. We also showcase the fact that the real-time extension of the semiempirical method INDO/S can be effectively applied for an approximate description of laser-driven dynamics in large systems.</p>\",\"PeriodicalId\":59,\"journal\":{\"name\":\"The Journal of Physical Chemistry A\",\"volume\":\"128 14\",\"pages\":\"2683–2702\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2024-03-27\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.acs.org/doi/epdf/10.1021/acs.jpca.3c07865\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry A\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jpca.3c07865\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry A","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpca.3c07865","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Modeling of High-Harmonic Generation in the C60 Fullerene Using Ab Initio, DFT-Based, and Semiempirical Methods
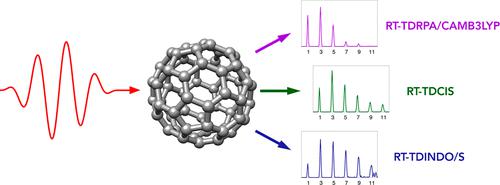
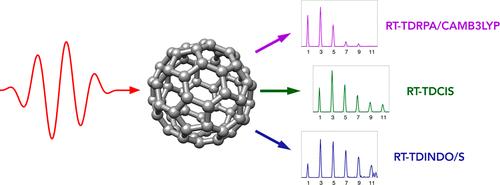
We report calculations of the high-harmonic generation spectra of the C60 fullerene molecule carried out by employing a diverse set of real-time time-dependent quantum chemical methods. All methodologies involve expanding the propagated electronic wave function in bases consisting of the ground and singly excited time-independent eigenstates obtained through the solution of the corresponding linear-response equations. We identify the correlation and exchange effect in the spectra by comparing the results from methods relying on the Hartree–Fock reference determinant with those obtained using approaches based on the density functional theory with different exchange–correlation functionals. The effect of the full random-phase approximation treatment of the excited electronic states is also analyzed and compared with the configuration interaction singles and the Tamm–Dancoff approximation. We also showcase the fact that the real-time extension of the semiempirical method INDO/S can be effectively applied for an approximate description of laser-driven dynamics in large systems.
期刊介绍:
The Journal of Physical Chemistry A is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


