Romdhane Rekaya, Shannon Smith, El Hamidi Hay, Nourhene Farhat, Samuel E Aggrey
{"title":"分析全基因组关联研究中具有特定结果误分类概率的二元反应。","authors":"Romdhane Rekaya, Shannon Smith, El Hamidi Hay, Nourhene Farhat, Samuel E Aggrey","doi":"10.2147/TACG.S122250","DOIUrl":null,"url":null,"abstract":"<p><p>Errors in the binary status of some response traits are frequent in human, animal, and plant applications. These error rates tend to differ between cases and controls because diagnostic and screening tests have different sensitivity and specificity. This increases the inaccuracies of classifying individuals into correct groups, giving rise to both false-positive and false-negative cases. The analysis of these noisy binary responses due to misclassification will undoubtedly reduce the statistical power of genome-wide association studies (GWAS). A threshold model that accommodates varying diagnostic errors between cases and controls was investigated. A simulation study was carried out where several binary data sets (case-control) were generated with varying effects for the most influential single nucleotide polymorphisms (SNPs) and different diagnostic error rate for cases and controls. Each simulated data set consisted of 2000 individuals. Ignoring misclassification resulted in biased estimates of true influential SNP effects and inflated estimates for true noninfluential markers. A substantial reduction in bias and increase in accuracy ranging from 12% to 32% was observed when the misclassification procedure was invoked. In fact, the majority of influential SNPs that were not identified using the noisy data were captured using the proposed method. Additionally, truly misclassified binary records were identified with high probability using the proposed method. The superiority of the proposed method was maintained across different simulation parameters (misclassification rates and odds ratios) attesting to its robustness.</p>","PeriodicalId":39131,"journal":{"name":"Application of Clinical Genetics","volume":"9 ","pages":"169-177"},"PeriodicalIF":2.6000,"publicationDate":"2016-11-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5138056/pdf/","citationCount":"0","resultStr":"{\"title\":\"Analysis of binary responses with outcome-specific misclassification probability in genome-wide association studies.\",\"authors\":\"Romdhane Rekaya, Shannon Smith, El Hamidi Hay, Nourhene Farhat, Samuel E Aggrey\",\"doi\":\"10.2147/TACG.S122250\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Errors in the binary status of some response traits are frequent in human, animal, and plant applications. These error rates tend to differ between cases and controls because diagnostic and screening tests have different sensitivity and specificity. This increases the inaccuracies of classifying individuals into correct groups, giving rise to both false-positive and false-negative cases. The analysis of these noisy binary responses due to misclassification will undoubtedly reduce the statistical power of genome-wide association studies (GWAS). A threshold model that accommodates varying diagnostic errors between cases and controls was investigated. A simulation study was carried out where several binary data sets (case-control) were generated with varying effects for the most influential single nucleotide polymorphisms (SNPs) and different diagnostic error rate for cases and controls. Each simulated data set consisted of 2000 individuals. Ignoring misclassification resulted in biased estimates of true influential SNP effects and inflated estimates for true noninfluential markers. A substantial reduction in bias and increase in accuracy ranging from 12% to 32% was observed when the misclassification procedure was invoked. In fact, the majority of influential SNPs that were not identified using the noisy data were captured using the proposed method. Additionally, truly misclassified binary records were identified with high probability using the proposed method. The superiority of the proposed method was maintained across different simulation parameters (misclassification rates and odds ratios) attesting to its robustness.</p>\",\"PeriodicalId\":39131,\"journal\":{\"name\":\"Application of Clinical Genetics\",\"volume\":\"9 \",\"pages\":\"169-177\"},\"PeriodicalIF\":2.6000,\"publicationDate\":\"2016-11-30\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5138056/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Application of Clinical Genetics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.2147/TACG.S122250\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2016/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Application of Clinical Genetics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.2147/TACG.S122250","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2016/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
摘要
在人类、动物和植物应用中,一些反应性状的二元状态经常出现错误。由于诊断和筛选测试具有不同的灵敏度和特异性,病例和对照之间的错误率往往不同。这增加了将个体归入正确组别的不准确性,导致假阳性和假阴性病例的出现。对这些因分类错误而产生的二元反应进行分析,无疑会降低全基因组关联研究(GWAS)的统计能力。我们研究了一种能适应病例和对照之间不同诊断误差的阈值模型。我们进行了一项模拟研究,生成了多个二元数据集(病例-对照),其中最有影响的单核苷酸多态性(SNPs)的效应各不相同,病例和对照的诊断错误率也各不相同。每个模拟数据集由 2000 个个体组成。忽略误分类会导致对真正有影响的 SNP 影响的估计值有偏差,而对真正无影响标记的估计值则会膨胀。当使用误分类程序时,偏差大幅减少,准确率提高了 12% 至 32%。事实上,使用所提出的方法可以捕捉到大部分未被噪声数据识别的有影响的 SNP。此外,使用所提出的方法,真正被错误分类的二进制记录被识别的概率也很高。拟议方法的优越性在不同的模拟参数(误分类率和几率)下都得以保持,这证明了它的稳健性。
Analysis of binary responses with outcome-specific misclassification probability in genome-wide association studies.
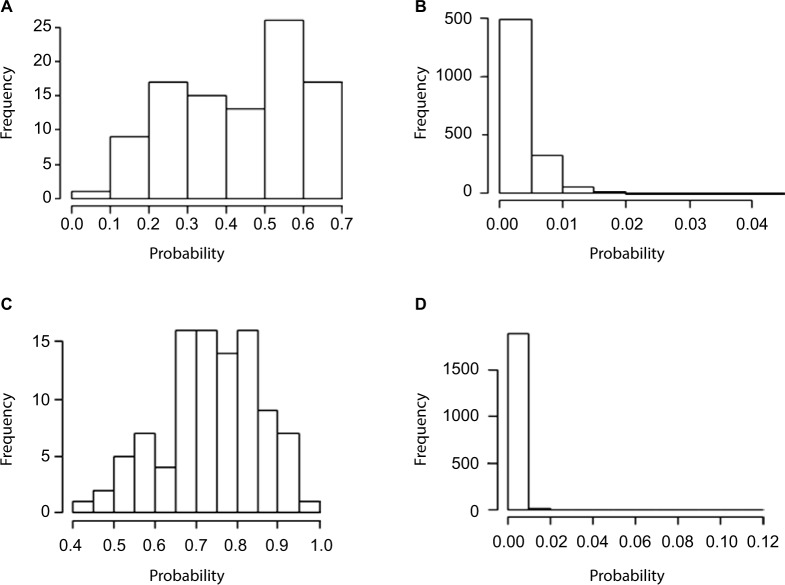
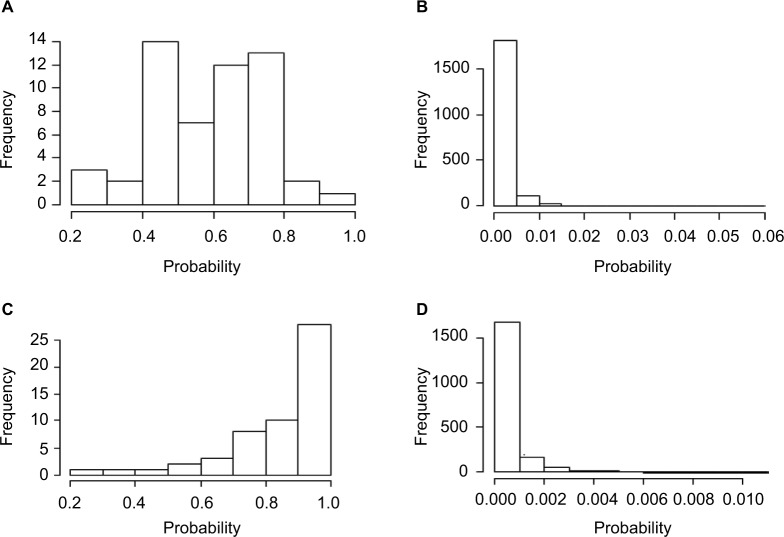
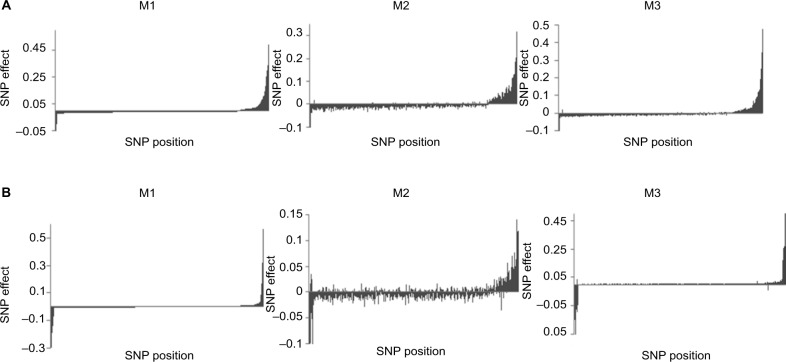
Errors in the binary status of some response traits are frequent in human, animal, and plant applications. These error rates tend to differ between cases and controls because diagnostic and screening tests have different sensitivity and specificity. This increases the inaccuracies of classifying individuals into correct groups, giving rise to both false-positive and false-negative cases. The analysis of these noisy binary responses due to misclassification will undoubtedly reduce the statistical power of genome-wide association studies (GWAS). A threshold model that accommodates varying diagnostic errors between cases and controls was investigated. A simulation study was carried out where several binary data sets (case-control) were generated with varying effects for the most influential single nucleotide polymorphisms (SNPs) and different diagnostic error rate for cases and controls. Each simulated data set consisted of 2000 individuals. Ignoring misclassification resulted in biased estimates of true influential SNP effects and inflated estimates for true noninfluential markers. A substantial reduction in bias and increase in accuracy ranging from 12% to 32% was observed when the misclassification procedure was invoked. In fact, the majority of influential SNPs that were not identified using the noisy data were captured using the proposed method. Additionally, truly misclassified binary records were identified with high probability using the proposed method. The superiority of the proposed method was maintained across different simulation parameters (misclassification rates and odds ratios) attesting to its robustness.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


