Mireia Seguí, Pedro Rodríguez-Jiménez, Aurora Fernández-Galván, Javier Fraga, Pablo Chicharro
{"title":"一名 48 岁女性身上的弥漫性色素沉着斑","authors":"Mireia Seguí, Pedro Rodríguez-Jiménez, Aurora Fernández-Galván, Javier Fraga, Pablo Chicharro","doi":"10.1002/jvc2.384","DOIUrl":null,"url":null,"abstract":"<p>A 48-year-old woman presented with a 4-year history of diffuse hyperpigmented papules beginning on the cleavage and progressing over the rest of the trunk and extremities. The lesions were mild pruritic. There were no family members with similar symptoms and both she and her family were from Spain and Caucasian. Her only significant past medical history was a papillary thyroid carcinoma that required total thyroidectomy. Clinical examination revealed generalized mottled hyper and hypopigmented macules on the trunk and extremities. Diffuse hyperpigmented small papules were also noted (Figure 1a−c). On dermoscopic examination, brownish areas with globular and dotted pigmentation alternating with round hypopigmented spots were observed (Figure 2). Laboratory investigations including a blood cell count, serum electrolytes and renal and liver function testing as well as protein serum electrophoresis revealed no abnormalities. Two skin biopsies were taken from the back of the patient (Figure 3).</p><p>Amyloidosis cutis dyschromica (ACD).</p><p>Two skin biopsies from the back showed deposition of amyloid in the papillary dermis confirmed by positive Congo red staining and immunohistochemical studies revealed that the amyloid expressed cytokeratins CK 5/6. There was also a mild inflammatory infiltrate, mostly composed of lymphocytes (Figure 3a,b). Based on the clinical and histopathological findings, the patient was diagnosed with ACD and due to the absence of symptoms scheduled for periodic follow-up.</p><p>ACD is a rare form of primary cutaneous amyloidosis, first described by Morishima in 1970.<span><sup>1</sup></span> ACD is characterized by (i) dotted, reticular hyperpigmentation with hypopigmented macules distributed over nearly all of the body, (ii) no or little itch, (iii) usual onset before puberty and (iv) focal amyloid deposition under the epidermis.<span><sup>1, 2</sup></span> Since this first description, both familiar and sporadic cases have been reported and most of the documented cases are from Asia. We herein present a sporadic case of a woman from Spain showing a late-onset of ACD.</p><p>Primary cutaneous amyloidosis is associated with the deposition of amyloid in the skin but not in internal organs. The most common variants of primary cutaneous amyloidosis include macular and lichen amyloidosis. ACD is a rare variant of primary cutaneous amyloidosis, characterized by reticular areas of hyperpigmentation with overlying hypopigmented macules and localized keratinocyte derived amyloid deposition within the papillary dermis. ACD has been most commonly reported in South and East Asian ethnic groups, having only few cases been reported in Caucasian ethnicity.<span><sup>2</sup></span></p><p>Although most of the reported cases are familiar, sporadic cases have also been reported. Recent studies point out that most cases of ACD result from autosomal recessive mutations in GPNMB, encoding glycoprotein nonmetastatic gene B.<span><sup>3, 4</sup></span> GPNMB has been found to have critical roles in melanosome formation, autophagy, phagocytosis, tissue repair and downregulation of inflammation. Nevertheless, the mechanism by which loss of function of GPNMB induces the pigment dyschromia and amyloidosis in ACD is not yet clear.</p><p>The onset of ACD is typically in early childhood or at a prepubertal age.<span><sup>2</sup></span> Our patient refers starting with the lesions at her forties, showing an unusual presentation of ACD. There are few cases of ACD with postpuberal onset reported in the literature.<span><sup>5</sup></span> The distinguishing features of the clinical presentation include generalized mottled hyper and hypopigmented macules. In addition, diffuse hyperpigmented small papules have also been documented. The size of the macules and papules range from 2 to 10 mm. The lesions are more pronounced on the trunk and the limbs with relative sparing of the face, hands, feet and neck. The extent of the dyspigmentation in ACD progresses very gradually over years, becoming more extensive through adulthood. Although the lesions of ACD are generally asymptomatic in opposition to the other subtypes of primary cutaneous amyloidosis, pruritus is reported in 19% of cases.<span><sup>2</sup></span></p><p>Histopathological findings include amyloid deposition in the papillary dermis confirmed by positive Congo red staining. A mild inflammatory infiltrate, mostly composed of lymphocytes and sparse melanophages, can also be found in the dermis. Positive staining for the cytokeratins CK34βE12 and CK5/6 in ACD suggests that the amyloid is derived from keratinocytes.<span><sup>6</sup></span> In our case, immunohistochemical studies were performed for cytokeratins CK 5/6, with positive staining.</p><p>The differential diagnosis of ACD includes dyschromatosis universalis hereditaria, xeroderma pigmentosum, poikiloderma-like amyloidosis, idiopathic guttate hypomelanosis and progressive macular hypomelanosis.</p><p>Treatment of ACD is not well documented, although acitretin has been reported to improve the dyschromia in some cases.</p><p>All authors discussed the results and contributed to the final manuscript.</p><p>The authors declare no conflict of interest.</p><p>The patient in this manuscript has given written informed consent for the use of her deidentified, anonymized, aggregated data and her case details (including photographs) for publication. Ethical approval: not applicable.</p>","PeriodicalId":94325,"journal":{"name":"JEADV clinical practice","volume":"3 2","pages":"782-784"},"PeriodicalIF":0.0000,"publicationDate":"2024-03-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jvc2.384","citationCount":"0","resultStr":"{\"title\":\"Diffuse hyperpigmented macules in a 48-year-old woman\",\"authors\":\"Mireia Seguí, Pedro Rodríguez-Jiménez, Aurora Fernández-Galván, Javier Fraga, Pablo Chicharro\",\"doi\":\"10.1002/jvc2.384\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>A 48-year-old woman presented with a 4-year history of diffuse hyperpigmented papules beginning on the cleavage and progressing over the rest of the trunk and extremities. The lesions were mild pruritic. There were no family members with similar symptoms and both she and her family were from Spain and Caucasian. Her only significant past medical history was a papillary thyroid carcinoma that required total thyroidectomy. Clinical examination revealed generalized mottled hyper and hypopigmented macules on the trunk and extremities. Diffuse hyperpigmented small papules were also noted (Figure 1a−c). On dermoscopic examination, brownish areas with globular and dotted pigmentation alternating with round hypopigmented spots were observed (Figure 2). Laboratory investigations including a blood cell count, serum electrolytes and renal and liver function testing as well as protein serum electrophoresis revealed no abnormalities. Two skin biopsies were taken from the back of the patient (Figure 3).</p><p>Amyloidosis cutis dyschromica (ACD).</p><p>Two skin biopsies from the back showed deposition of amyloid in the papillary dermis confirmed by positive Congo red staining and immunohistochemical studies revealed that the amyloid expressed cytokeratins CK 5/6. There was also a mild inflammatory infiltrate, mostly composed of lymphocytes (Figure 3a,b). Based on the clinical and histopathological findings, the patient was diagnosed with ACD and due to the absence of symptoms scheduled for periodic follow-up.</p><p>ACD is a rare form of primary cutaneous amyloidosis, first described by Morishima in 1970.<span><sup>1</sup></span> ACD is characterized by (i) dotted, reticular hyperpigmentation with hypopigmented macules distributed over nearly all of the body, (ii) no or little itch, (iii) usual onset before puberty and (iv) focal amyloid deposition under the epidermis.<span><sup>1, 2</sup></span> Since this first description, both familiar and sporadic cases have been reported and most of the documented cases are from Asia. We herein present a sporadic case of a woman from Spain showing a late-onset of ACD.</p><p>Primary cutaneous amyloidosis is associated with the deposition of amyloid in the skin but not in internal organs. The most common variants of primary cutaneous amyloidosis include macular and lichen amyloidosis. ACD is a rare variant of primary cutaneous amyloidosis, characterized by reticular areas of hyperpigmentation with overlying hypopigmented macules and localized keratinocyte derived amyloid deposition within the papillary dermis. ACD has been most commonly reported in South and East Asian ethnic groups, having only few cases been reported in Caucasian ethnicity.<span><sup>2</sup></span></p><p>Although most of the reported cases are familiar, sporadic cases have also been reported. Recent studies point out that most cases of ACD result from autosomal recessive mutations in GPNMB, encoding glycoprotein nonmetastatic gene B.<span><sup>3, 4</sup></span> GPNMB has been found to have critical roles in melanosome formation, autophagy, phagocytosis, tissue repair and downregulation of inflammation. Nevertheless, the mechanism by which loss of function of GPNMB induces the pigment dyschromia and amyloidosis in ACD is not yet clear.</p><p>The onset of ACD is typically in early childhood or at a prepubertal age.<span><sup>2</sup></span> Our patient refers starting with the lesions at her forties, showing an unusual presentation of ACD. There are few cases of ACD with postpuberal onset reported in the literature.<span><sup>5</sup></span> The distinguishing features of the clinical presentation include generalized mottled hyper and hypopigmented macules. In addition, diffuse hyperpigmented small papules have also been documented. The size of the macules and papules range from 2 to 10 mm. The lesions are more pronounced on the trunk and the limbs with relative sparing of the face, hands, feet and neck. The extent of the dyspigmentation in ACD progresses very gradually over years, becoming more extensive through adulthood. Although the lesions of ACD are generally asymptomatic in opposition to the other subtypes of primary cutaneous amyloidosis, pruritus is reported in 19% of cases.<span><sup>2</sup></span></p><p>Histopathological findings include amyloid deposition in the papillary dermis confirmed by positive Congo red staining. A mild inflammatory infiltrate, mostly composed of lymphocytes and sparse melanophages, can also be found in the dermis. Positive staining for the cytokeratins CK34βE12 and CK5/6 in ACD suggests that the amyloid is derived from keratinocytes.<span><sup>6</sup></span> In our case, immunohistochemical studies were performed for cytokeratins CK 5/6, with positive staining.</p><p>The differential diagnosis of ACD includes dyschromatosis universalis hereditaria, xeroderma pigmentosum, poikiloderma-like amyloidosis, idiopathic guttate hypomelanosis and progressive macular hypomelanosis.</p><p>Treatment of ACD is not well documented, although acitretin has been reported to improve the dyschromia in some cases.</p><p>All authors discussed the results and contributed to the final manuscript.</p><p>The authors declare no conflict of interest.</p><p>The patient in this manuscript has given written informed consent for the use of her deidentified, anonymized, aggregated data and her case details (including photographs) for publication. Ethical approval: not applicable.</p>\",\"PeriodicalId\":94325,\"journal\":{\"name\":\"JEADV clinical practice\",\"volume\":\"3 2\",\"pages\":\"782-784\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2024-03-19\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jvc2.384\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"JEADV clinical practice\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jvc2.384\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"JEADV clinical practice","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jvc2.384","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
Diffuse hyperpigmented macules in a 48-year-old woman
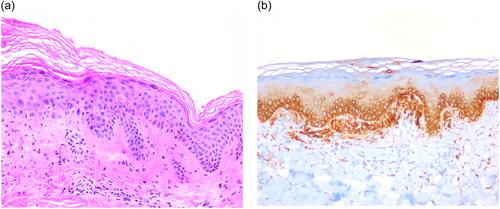
A 48-year-old woman presented with a 4-year history of diffuse hyperpigmented papules beginning on the cleavage and progressing over the rest of the trunk and extremities. The lesions were mild pruritic. There were no family members with similar symptoms and both she and her family were from Spain and Caucasian. Her only significant past medical history was a papillary thyroid carcinoma that required total thyroidectomy. Clinical examination revealed generalized mottled hyper and hypopigmented macules on the trunk and extremities. Diffuse hyperpigmented small papules were also noted (Figure 1a−c). On dermoscopic examination, brownish areas with globular and dotted pigmentation alternating with round hypopigmented spots were observed (Figure 2). Laboratory investigations including a blood cell count, serum electrolytes and renal and liver function testing as well as protein serum electrophoresis revealed no abnormalities. Two skin biopsies were taken from the back of the patient (Figure 3).
Amyloidosis cutis dyschromica (ACD).
Two skin biopsies from the back showed deposition of amyloid in the papillary dermis confirmed by positive Congo red staining and immunohistochemical studies revealed that the amyloid expressed cytokeratins CK 5/6. There was also a mild inflammatory infiltrate, mostly composed of lymphocytes (Figure 3a,b). Based on the clinical and histopathological findings, the patient was diagnosed with ACD and due to the absence of symptoms scheduled for periodic follow-up.
ACD is a rare form of primary cutaneous amyloidosis, first described by Morishima in 1970.1 ACD is characterized by (i) dotted, reticular hyperpigmentation with hypopigmented macules distributed over nearly all of the body, (ii) no or little itch, (iii) usual onset before puberty and (iv) focal amyloid deposition under the epidermis.1, 2 Since this first description, both familiar and sporadic cases have been reported and most of the documented cases are from Asia. We herein present a sporadic case of a woman from Spain showing a late-onset of ACD.
Primary cutaneous amyloidosis is associated with the deposition of amyloid in the skin but not in internal organs. The most common variants of primary cutaneous amyloidosis include macular and lichen amyloidosis. ACD is a rare variant of primary cutaneous amyloidosis, characterized by reticular areas of hyperpigmentation with overlying hypopigmented macules and localized keratinocyte derived amyloid deposition within the papillary dermis. ACD has been most commonly reported in South and East Asian ethnic groups, having only few cases been reported in Caucasian ethnicity.2
Although most of the reported cases are familiar, sporadic cases have also been reported. Recent studies point out that most cases of ACD result from autosomal recessive mutations in GPNMB, encoding glycoprotein nonmetastatic gene B.3, 4 GPNMB has been found to have critical roles in melanosome formation, autophagy, phagocytosis, tissue repair and downregulation of inflammation. Nevertheless, the mechanism by which loss of function of GPNMB induces the pigment dyschromia and amyloidosis in ACD is not yet clear.
The onset of ACD is typically in early childhood or at a prepubertal age.2 Our patient refers starting with the lesions at her forties, showing an unusual presentation of ACD. There are few cases of ACD with postpuberal onset reported in the literature.5 The distinguishing features of the clinical presentation include generalized mottled hyper and hypopigmented macules. In addition, diffuse hyperpigmented small papules have also been documented. The size of the macules and papules range from 2 to 10 mm. The lesions are more pronounced on the trunk and the limbs with relative sparing of the face, hands, feet and neck. The extent of the dyspigmentation in ACD progresses very gradually over years, becoming more extensive through adulthood. Although the lesions of ACD are generally asymptomatic in opposition to the other subtypes of primary cutaneous amyloidosis, pruritus is reported in 19% of cases.2
Histopathological findings include amyloid deposition in the papillary dermis confirmed by positive Congo red staining. A mild inflammatory infiltrate, mostly composed of lymphocytes and sparse melanophages, can also be found in the dermis. Positive staining for the cytokeratins CK34βE12 and CK5/6 in ACD suggests that the amyloid is derived from keratinocytes.6 In our case, immunohistochemical studies were performed for cytokeratins CK 5/6, with positive staining.
The differential diagnosis of ACD includes dyschromatosis universalis hereditaria, xeroderma pigmentosum, poikiloderma-like amyloidosis, idiopathic guttate hypomelanosis and progressive macular hypomelanosis.
Treatment of ACD is not well documented, although acitretin has been reported to improve the dyschromia in some cases.
All authors discussed the results and contributed to the final manuscript.
The authors declare no conflict of interest.
The patient in this manuscript has given written informed consent for the use of her deidentified, anonymized, aggregated data and her case details (including photographs) for publication. Ethical approval: not applicable.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


