D. I. Zhigalina, T. N. Kireeva, T. V. Nikitina, O. N. Odinokova, N. A. Kolesnikov, A. A. Malakhova, R. R. Savchenko, I. Zh. Zhalsanova, N. R. Valiahmetov, A. E. Postrigan, S. L. Vovk, N. B. Torkhova, A. A. Frolova, V. A. Stepanov, N. A. Skryabin
{"title":"通过重编程一名同型 F508del 囊性纤维化患者的成纤维细胞,生成诱导多能干细胞系 TNRMCi001-A","authors":"D. I. Zhigalina, T. N. Kireeva, T. V. Nikitina, O. N. Odinokova, N. A. Kolesnikov, A. A. Malakhova, R. R. Savchenko, I. Zh. Zhalsanova, N. R. Valiahmetov, A. E. Postrigan, S. L. Vovk, N. B. Torkhova, A. A. Frolova, V. A. Stepanov, N. A. Skryabin","doi":"10.1134/s106236042307007x","DOIUrl":null,"url":null,"abstract":"<h3 data-test=\"abstract-sub-heading\">Abstract</h3><p>Cystic fibrosis (CF) is a hereditary disease that leads to impaired functioning of chloride channels in cells, and, as a result, to a decrease in the viscoelastic properties of the secretion of all exocrine glands. Cystic fibrosis is the result of mutations in the cystic fibrosis transmembrane conductance regulator (<i>CFTR</i>) gene, which encodes the CFTR protein. In this study, induced pluripotent stem cells (iPSCs) were obtained from the skin fibroblasts of a patient with a homozygous mutation F508del <i>CFTR</i> (NM_000492.3(CFTR):c.1521_1523del). This deletion is the most common for cystic fibrosis. The resulting iPSC line had a normal karyotype, retained the original genotype, and also demonstrated the presence of pluripotency markers (OCT4, SOX2, NANOG, SSEA4, TRA-1-60) and the ability to differentiate into derivatives of three germ layers.</p>","PeriodicalId":21434,"journal":{"name":"Russian Journal of Developmental Biology","volume":"30 1","pages":""},"PeriodicalIF":0.4000,"publicationDate":"2024-03-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Generation of an Induced Pluripotent Stem Cell Line TNRMCi001-A by Reprogramming Fibroblasts from a Homozygous F508del Cystic Fibrosis Patient\",\"authors\":\"D. I. Zhigalina, T. N. Kireeva, T. V. Nikitina, O. N. Odinokova, N. A. Kolesnikov, A. A. Malakhova, R. R. Savchenko, I. Zh. Zhalsanova, N. R. Valiahmetov, A. E. Postrigan, S. L. Vovk, N. B. Torkhova, A. A. Frolova, V. A. Stepanov, N. A. Skryabin\",\"doi\":\"10.1134/s106236042307007x\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<h3 data-test=\\\"abstract-sub-heading\\\">Abstract</h3><p>Cystic fibrosis (CF) is a hereditary disease that leads to impaired functioning of chloride channels in cells, and, as a result, to a decrease in the viscoelastic properties of the secretion of all exocrine glands. Cystic fibrosis is the result of mutations in the cystic fibrosis transmembrane conductance regulator (<i>CFTR</i>) gene, which encodes the CFTR protein. In this study, induced pluripotent stem cells (iPSCs) were obtained from the skin fibroblasts of a patient with a homozygous mutation F508del <i>CFTR</i> (NM_000492.3(CFTR):c.1521_1523del). This deletion is the most common for cystic fibrosis. The resulting iPSC line had a normal karyotype, retained the original genotype, and also demonstrated the presence of pluripotency markers (OCT4, SOX2, NANOG, SSEA4, TRA-1-60) and the ability to differentiate into derivatives of three germ layers.</p>\",\"PeriodicalId\":21434,\"journal\":{\"name\":\"Russian Journal of Developmental Biology\",\"volume\":\"30 1\",\"pages\":\"\"},\"PeriodicalIF\":0.4000,\"publicationDate\":\"2024-03-23\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Russian Journal of Developmental Biology\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1134/s106236042307007x\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"DEVELOPMENTAL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Russian Journal of Developmental Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1134/s106236042307007x","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"DEVELOPMENTAL BIOLOGY","Score":null,"Total":0}
Generation of an Induced Pluripotent Stem Cell Line TNRMCi001-A by Reprogramming Fibroblasts from a Homozygous F508del Cystic Fibrosis Patient
Abstract
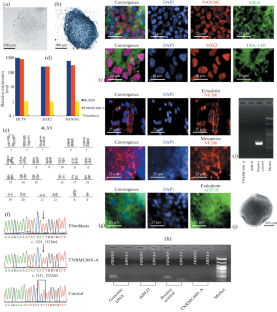
Cystic fibrosis (CF) is a hereditary disease that leads to impaired functioning of chloride channels in cells, and, as a result, to a decrease in the viscoelastic properties of the secretion of all exocrine glands. Cystic fibrosis is the result of mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, which encodes the CFTR protein. In this study, induced pluripotent stem cells (iPSCs) were obtained from the skin fibroblasts of a patient with a homozygous mutation F508del CFTR (NM_000492.3(CFTR):c.1521_1523del). This deletion is the most common for cystic fibrosis. The resulting iPSC line had a normal karyotype, retained the original genotype, and also demonstrated the presence of pluripotency markers (OCT4, SOX2, NANOG, SSEA4, TRA-1-60) and the ability to differentiate into derivatives of three germ layers.
期刊介绍:
Russian Journal of Developmental Biology is a journal that publishes reviews and experimental articles dealing with various aspects of developmental biology. It presents fundamental and applied research on development, regeneration, and carcinogenesis at the molecular, cellular, and organismic levels.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


