Francis D. Phimister, Rachel C. Anderson, David G. Thomas, Michelle J. Farquhar, Paul Maclean, Ruy Jauregui, Wayne Young, Christina F. Butowski, Emma N. Bermingham
{"title":"利用荟萃分析了解膳食蛋白质和脂肪含量对家犬粪便微生物群组成的影响:一项试点研究。","authors":"Francis D. Phimister, Rachel C. Anderson, David G. Thomas, Michelle J. Farquhar, Paul Maclean, Ruy Jauregui, Wayne Young, Christina F. Butowski, Emma N. Bermingham","doi":"10.1002/mbo3.1404","DOIUrl":null,"url":null,"abstract":"<p>The interplay between diet and fecal microbiota composition is garnering increased interest across various host species, including domestic dogs. While the influence of dietary macronutrients and their associated microbial communities have been extensively reviewed, these reviews are descriptive and do not account for differences in microbial community analysis, nor do they standardize macronutrient content across studies. To address this, a meta-analysis was performed to assess the impact of dietary crude protein (“protein”) and dietary crude fat (“fat”) on the fecal microbiota composition in healthy dogs. Sixteen publications met the eligibility criteria for the meta-analysis, yielding a final data set of 314 dogs. Diets were classed as low, moderate, high, or supra in terms of protein or fat content. Sequence data from each publication were retrieved from public databases and reanalyzed using consistent bioinformatic pipelines. Analysis of community diversity indices and unsupervised clustering of the data with principal coordinate analysis revealed a small effect size and complete overlap between protein and fat levels at the overall community level. Supervised clustering through random forest analysis and partial least squares-discriminant analysis indicated alterations in the fecal microbiota composition at a more individual taxonomic level, corresponding to the levels of protein or fat. The <i>Prevotellaceae Ga6A1 group</i> and <i>Enterococcus</i> were associated with increasing levels of protein, while <i>Allobaculum</i> and <i>Clostridium sensu stricto 13</i> were associated with increasing levels of fat. Interestingly, the random forest analyses revealed that <i>Sharpea</i>, despite its low relative abundance in the dog's fecal microbiome, was primarily responsible for the separation of the microbiome for both protein and fat. Future research should focus on validating and understanding the functional roles of these relatively low-abundant genera.</p>","PeriodicalId":18573,"journal":{"name":"MicrobiologyOpen","volume":"13 2","pages":""},"PeriodicalIF":4.6000,"publicationDate":"2024-03-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/mbo3.1404","citationCount":"0","resultStr":"{\"title\":\"Using meta-analysis to understand the impacts of dietary protein and fat content on the composition of fecal microbiota of domestic dogs (Canis lupus familiaris): A pilot study\",\"authors\":\"Francis D. Phimister, Rachel C. Anderson, David G. Thomas, Michelle J. Farquhar, Paul Maclean, Ruy Jauregui, Wayne Young, Christina F. Butowski, Emma N. Bermingham\",\"doi\":\"10.1002/mbo3.1404\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>The interplay between diet and fecal microbiota composition is garnering increased interest across various host species, including domestic dogs. While the influence of dietary macronutrients and their associated microbial communities have been extensively reviewed, these reviews are descriptive and do not account for differences in microbial community analysis, nor do they standardize macronutrient content across studies. To address this, a meta-analysis was performed to assess the impact of dietary crude protein (“protein”) and dietary crude fat (“fat”) on the fecal microbiota composition in healthy dogs. Sixteen publications met the eligibility criteria for the meta-analysis, yielding a final data set of 314 dogs. Diets were classed as low, moderate, high, or supra in terms of protein or fat content. Sequence data from each publication were retrieved from public databases and reanalyzed using consistent bioinformatic pipelines. Analysis of community diversity indices and unsupervised clustering of the data with principal coordinate analysis revealed a small effect size and complete overlap between protein and fat levels at the overall community level. Supervised clustering through random forest analysis and partial least squares-discriminant analysis indicated alterations in the fecal microbiota composition at a more individual taxonomic level, corresponding to the levels of protein or fat. The <i>Prevotellaceae Ga6A1 group</i> and <i>Enterococcus</i> were associated with increasing levels of protein, while <i>Allobaculum</i> and <i>Clostridium sensu stricto 13</i> were associated with increasing levels of fat. Interestingly, the random forest analyses revealed that <i>Sharpea</i>, despite its low relative abundance in the dog's fecal microbiome, was primarily responsible for the separation of the microbiome for both protein and fat. Future research should focus on validating and understanding the functional roles of these relatively low-abundant genera.</p>\",\"PeriodicalId\":18573,\"journal\":{\"name\":\"MicrobiologyOpen\",\"volume\":\"13 2\",\"pages\":\"\"},\"PeriodicalIF\":4.6000,\"publicationDate\":\"2024-03-21\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/mbo3.1404\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"MicrobiologyOpen\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/mbo3.1404\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"MICROBIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"MicrobiologyOpen","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/mbo3.1404","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"MICROBIOLOGY","Score":null,"Total":0}
Using meta-analysis to understand the impacts of dietary protein and fat content on the composition of fecal microbiota of domestic dogs (Canis lupus familiaris): A pilot study
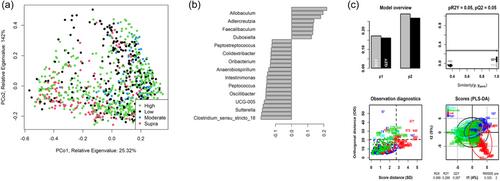
The interplay between diet and fecal microbiota composition is garnering increased interest across various host species, including domestic dogs. While the influence of dietary macronutrients and their associated microbial communities have been extensively reviewed, these reviews are descriptive and do not account for differences in microbial community analysis, nor do they standardize macronutrient content across studies. To address this, a meta-analysis was performed to assess the impact of dietary crude protein (“protein”) and dietary crude fat (“fat”) on the fecal microbiota composition in healthy dogs. Sixteen publications met the eligibility criteria for the meta-analysis, yielding a final data set of 314 dogs. Diets were classed as low, moderate, high, or supra in terms of protein or fat content. Sequence data from each publication were retrieved from public databases and reanalyzed using consistent bioinformatic pipelines. Analysis of community diversity indices and unsupervised clustering of the data with principal coordinate analysis revealed a small effect size and complete overlap between protein and fat levels at the overall community level. Supervised clustering through random forest analysis and partial least squares-discriminant analysis indicated alterations in the fecal microbiota composition at a more individual taxonomic level, corresponding to the levels of protein or fat. The Prevotellaceae Ga6A1 group and Enterococcus were associated with increasing levels of protein, while Allobaculum and Clostridium sensu stricto 13 were associated with increasing levels of fat. Interestingly, the random forest analyses revealed that Sharpea, despite its low relative abundance in the dog's fecal microbiome, was primarily responsible for the separation of the microbiome for both protein and fat. Future research should focus on validating and understanding the functional roles of these relatively low-abundant genera.
期刊介绍:
MicrobiologyOpen is a peer reviewed, fully open access, broad-scope, and interdisciplinary journal delivering rapid decisions and fast publication of microbial science, a field which is undergoing a profound and exciting evolution in this post-genomic era.
The journal aims to serve the research community by providing a vehicle for authors wishing to publish quality research in both fundamental and applied microbiology. Our goal is to publish articles that stimulate discussion and debate, as well as add to our knowledge base and further the understanding of microbial interactions and microbial processes.
MicrobiologyOpen gives prompt and equal consideration to articles reporting theoretical, experimental, applied, and descriptive work in all aspects of bacteriology, virology, mycology and protistology, including, but not limited to:
- agriculture
- antimicrobial resistance
- astrobiology
- biochemistry
- biotechnology
- cell and molecular biology
- clinical microbiology
- computational, systems, and synthetic microbiology
- environmental science
- evolutionary biology, ecology, and systematics
- food science and technology
- genetics and genomics
- geobiology and earth science
- host-microbe interactions
- infectious diseases
- natural products discovery
- pharmaceutical and medicinal chemistry
- physiology
- plant pathology
- veterinary microbiology
We will consider submissions across unicellular and cell-cluster organisms: prokaryotes (bacteria, archaea) and eukaryotes (fungi, protists, microalgae, lichens), as well as viruses and prions infecting or interacting with microorganisms, plants and animals, including genetic, biochemical, biophysical, bioinformatic and structural analyses.
The journal features Original Articles (including full Research articles, Method articles, and Short Communications), Commentaries, Reviews, and Editorials. Original papers must report well-conducted research with conclusions supported by the data presented in the article. We also support confirmatory research and aim to work with authors to meet reviewer expectations.
MicrobiologyOpen publishes articles submitted directly to the journal and those referred from other Wiley journals.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


