Ho Heon Kim, Dong-Wook Kim, Junwoo Woo, Kyoungyeul Lee
{"title":"通过整合基于规则的算法和机器学习算法,对遗传变异进行可解释的优先排序,以诊断罕见孟德尔疾病。","authors":"Ho Heon Kim, Dong-Wook Kim, Junwoo Woo, Kyoungyeul Lee","doi":"10.1186/s40246-024-00595-8","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>In the process of finding the causative variant of rare diseases, accurate assessment and prioritization of genetic variants is essential. Previous variant prioritization tools mainly depend on the in-silico prediction of the pathogenicity of variants, which results in low sensitivity and difficulty in interpreting the prioritization result. In this study, we propose an explainable algorithm for variant prioritization, named 3ASC, with higher sensitivity and ability to annotate evidence used for prioritization. 3ASC annotates each variant with the 28 criteria defined by the ACMG/AMP genome interpretation guidelines and features related to the clinical interpretation of the variants. The system can explain the result based on annotated evidence and feature contributions.</p><p><strong>Results: </strong>We trained various machine learning algorithms using in-house patient data. The performance of variant ranking was assessed using the recall rate of identifying causative variants in the top-ranked variants. The best practice model was a random forest classifier that showed top 1 recall of 85.6% and top 3 recall of 94.4%. The 3ASC annotates the ACMG/AMP criteria for each genetic variant of a patient so that clinical geneticists can interpret the result as in the CAGI6 SickKids challenge. In the challenge, 3ASC identified causal genes for 10 out of 14 patient cases, with evidence of decreased gene expression for 6 cases. Among them, two genes (HDAC8 and CASK) had decreased gene expression profiles confirmed by transcriptome data.</p><p><strong>Conclusions: </strong>3ASC can prioritize genetic variants with higher sensitivity compared to previous methods by integrating various features related to clinical interpretation, including features related to false positive risk such as quality control and disease inheritance pattern. The system allows interpretation of each variant based on the ACMG/AMP criteria and feature contribution assessed using explainable AI techniques.</p>","PeriodicalId":13183,"journal":{"name":"Human Genomics","volume":"18 1","pages":"28"},"PeriodicalIF":4.3000,"publicationDate":"2024-03-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10956189/pdf/","citationCount":"0","resultStr":"{\"title\":\"Explicable prioritization of genetic variants by integration of rule-based and machine learning algorithms for diagnosis of rare Mendelian disorders.\",\"authors\":\"Ho Heon Kim, Dong-Wook Kim, Junwoo Woo, Kyoungyeul Lee\",\"doi\":\"10.1186/s40246-024-00595-8\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>In the process of finding the causative variant of rare diseases, accurate assessment and prioritization of genetic variants is essential. Previous variant prioritization tools mainly depend on the in-silico prediction of the pathogenicity of variants, which results in low sensitivity and difficulty in interpreting the prioritization result. In this study, we propose an explainable algorithm for variant prioritization, named 3ASC, with higher sensitivity and ability to annotate evidence used for prioritization. 3ASC annotates each variant with the 28 criteria defined by the ACMG/AMP genome interpretation guidelines and features related to the clinical interpretation of the variants. The system can explain the result based on annotated evidence and feature contributions.</p><p><strong>Results: </strong>We trained various machine learning algorithms using in-house patient data. The performance of variant ranking was assessed using the recall rate of identifying causative variants in the top-ranked variants. The best practice model was a random forest classifier that showed top 1 recall of 85.6% and top 3 recall of 94.4%. The 3ASC annotates the ACMG/AMP criteria for each genetic variant of a patient so that clinical geneticists can interpret the result as in the CAGI6 SickKids challenge. In the challenge, 3ASC identified causal genes for 10 out of 14 patient cases, with evidence of decreased gene expression for 6 cases. Among them, two genes (HDAC8 and CASK) had decreased gene expression profiles confirmed by transcriptome data.</p><p><strong>Conclusions: </strong>3ASC can prioritize genetic variants with higher sensitivity compared to previous methods by integrating various features related to clinical interpretation, including features related to false positive risk such as quality control and disease inheritance pattern. The system allows interpretation of each variant based on the ACMG/AMP criteria and feature contribution assessed using explainable AI techniques.</p>\",\"PeriodicalId\":13183,\"journal\":{\"name\":\"Human Genomics\",\"volume\":\"18 1\",\"pages\":\"28\"},\"PeriodicalIF\":4.3000,\"publicationDate\":\"2024-03-21\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10956189/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Human Genomics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s40246-024-00595-8\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human Genomics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s40246-024-00595-8","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Explicable prioritization of genetic variants by integration of rule-based and machine learning algorithms for diagnosis of rare Mendelian disorders.
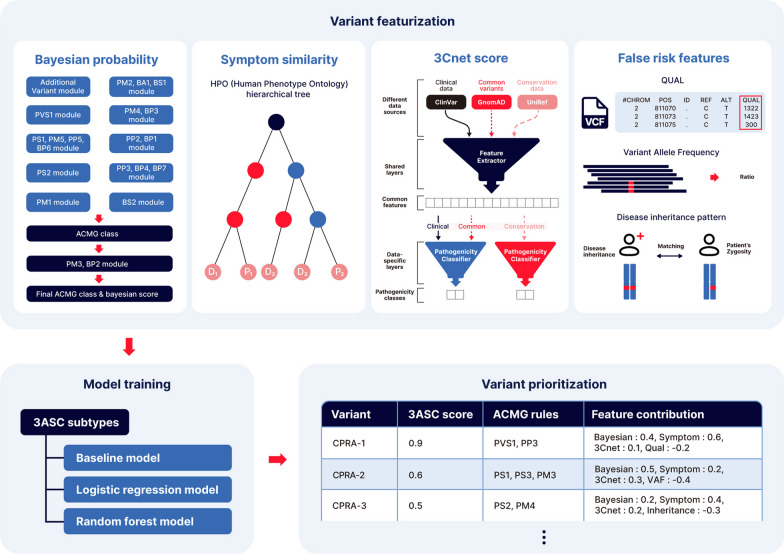
Background: In the process of finding the causative variant of rare diseases, accurate assessment and prioritization of genetic variants is essential. Previous variant prioritization tools mainly depend on the in-silico prediction of the pathogenicity of variants, which results in low sensitivity and difficulty in interpreting the prioritization result. In this study, we propose an explainable algorithm for variant prioritization, named 3ASC, with higher sensitivity and ability to annotate evidence used for prioritization. 3ASC annotates each variant with the 28 criteria defined by the ACMG/AMP genome interpretation guidelines and features related to the clinical interpretation of the variants. The system can explain the result based on annotated evidence and feature contributions.
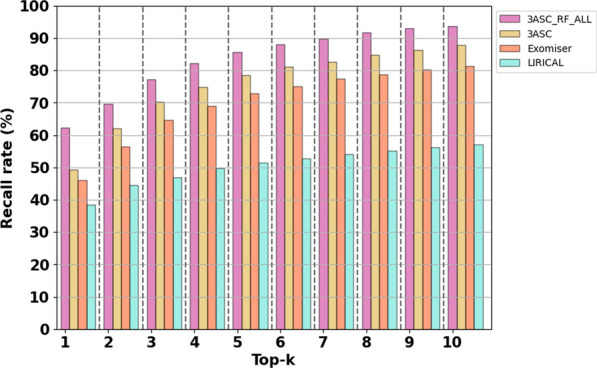
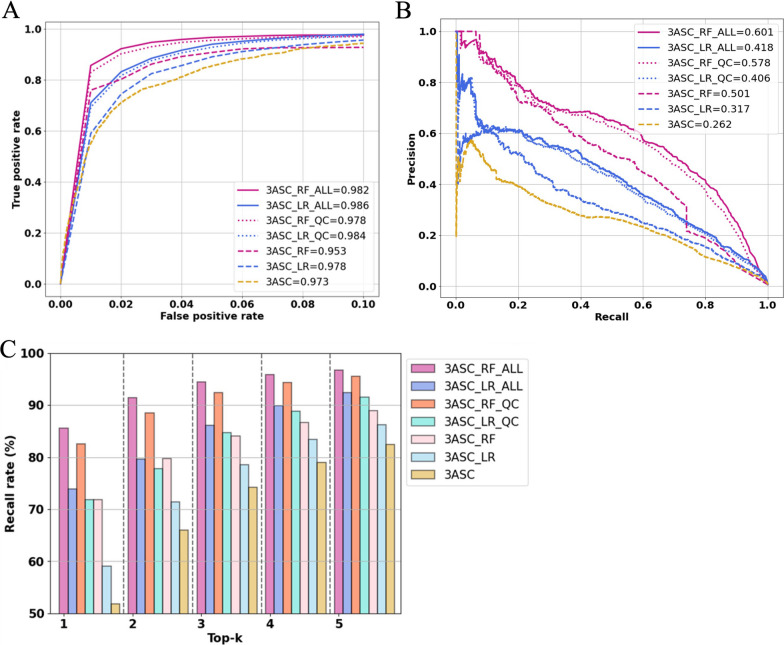
Results: We trained various machine learning algorithms using in-house patient data. The performance of variant ranking was assessed using the recall rate of identifying causative variants in the top-ranked variants. The best practice model was a random forest classifier that showed top 1 recall of 85.6% and top 3 recall of 94.4%. The 3ASC annotates the ACMG/AMP criteria for each genetic variant of a patient so that clinical geneticists can interpret the result as in the CAGI6 SickKids challenge. In the challenge, 3ASC identified causal genes for 10 out of 14 patient cases, with evidence of decreased gene expression for 6 cases. Among them, two genes (HDAC8 and CASK) had decreased gene expression profiles confirmed by transcriptome data.
Conclusions: 3ASC can prioritize genetic variants with higher sensitivity compared to previous methods by integrating various features related to clinical interpretation, including features related to false positive risk such as quality control and disease inheritance pattern. The system allows interpretation of each variant based on the ACMG/AMP criteria and feature contribution assessed using explainable AI techniques.
期刊介绍:
Human Genomics is a peer-reviewed, open access, online journal that focuses on the application of genomic analysis in all aspects of human health and disease, as well as genomic analysis of drug efficacy and safety, and comparative genomics.
Topics covered by the journal include, but are not limited to: pharmacogenomics, genome-wide association studies, genome-wide sequencing, exome sequencing, next-generation deep-sequencing, functional genomics, epigenomics, translational genomics, expression profiling, proteomics, bioinformatics, animal models, statistical genetics, genetic epidemiology, human population genetics and comparative genomics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


