Benoit Mazel, Julian Delanne, Aurore Garde, Caroline Racine, Ange-Line Bruel, Yannis Duffourd, Diego Lopergolo, Filippo Maria Santorelli, Viviana Marchi, Anna Maria Pinto, Maria Antonietta Mencarelli, Roberto Canitano, Floriana Valentino, Filomena Tiziana Papa, Chiara Fallerini, Francesca Mari, Alessandra Renieri, Arnold Munnich, Tanguy Niclass, Gwenaël Le Guyader, Christel Thauvin-Robinet, Christophe Philippe, Laurence Faivre
{"title":"与先天性雷特综合征相比,FOXG1变体可能与较轻的表型有关,即无助行走和语言发育。","authors":"Benoit Mazel, Julian Delanne, Aurore Garde, Caroline Racine, Ange-Line Bruel, Yannis Duffourd, Diego Lopergolo, Filippo Maria Santorelli, Viviana Marchi, Anna Maria Pinto, Maria Antonietta Mencarelli, Roberto Canitano, Floriana Valentino, Filomena Tiziana Papa, Chiara Fallerini, Francesca Mari, Alessandra Renieri, Arnold Munnich, Tanguy Niclass, Gwenaël Le Guyader, Christel Thauvin-Robinet, Christophe Philippe, Laurence Faivre","doi":"10.1002/ajmg.b.32970","DOIUrl":null,"url":null,"abstract":"<p>Since 2008, <i>FOXG1</i> haploinsufficiency has been linked to a severe neurodevelopmental phenotype resembling Rett syndrome but with earlier onset. Most patients are unable to sit, walk, or speak. For years, <i>FOXG1</i> sequencing was only prescribed in such severe cases, limiting insight into the full clinical spectrum associated with this gene. Next-generation sequencing (NGS) now enables unbiased diagnostics. Through the European Reference Network for Rare Malformation Syndromes, Intellectual and Other Neurodevelopmental Disorders, we gathered data from patients with heterozygous <i>FOXG1</i> variants presenting a mild phenotype, defined as able to speak and walk independently. We also reviewed data from three previously reported patients meeting our criteria. We identified five new patients with pathogenic <i>FOXG1</i> missense variants, primarily in the forkhead domain, showing varying nonspecific intellectual disability and developmental delay. These features are not typical of congenital Rett syndrome and were rarely associated with microcephaly and epilepsy. Our findings are consistent with a previous genotype–phenotype analysis by Mitter et al. suggesting the delineation of five different <i>FOXG1</i> genotype groups. Milder phenotypes were associated with missense variants in the forkhead domain. This information may facilitate prognostic assessments in children carrying a <i>FOXG1</i> variant and improve the interpretation of new variants identified with genomic sequencing.</p>","PeriodicalId":7673,"journal":{"name":"American Journal of Medical Genetics Part B: Neuropsychiatric Genetics","volume":"195 6","pages":""},"PeriodicalIF":1.5000,"publicationDate":"2024-03-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/ajmg.b.32970","citationCount":"0","resultStr":"{\"title\":\"FOXG1 variants can be associated with milder phenotypes than congenital Rett syndrome with unassisted walking and language development\",\"authors\":\"Benoit Mazel, Julian Delanne, Aurore Garde, Caroline Racine, Ange-Line Bruel, Yannis Duffourd, Diego Lopergolo, Filippo Maria Santorelli, Viviana Marchi, Anna Maria Pinto, Maria Antonietta Mencarelli, Roberto Canitano, Floriana Valentino, Filomena Tiziana Papa, Chiara Fallerini, Francesca Mari, Alessandra Renieri, Arnold Munnich, Tanguy Niclass, Gwenaël Le Guyader, Christel Thauvin-Robinet, Christophe Philippe, Laurence Faivre\",\"doi\":\"10.1002/ajmg.b.32970\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Since 2008, <i>FOXG1</i> haploinsufficiency has been linked to a severe neurodevelopmental phenotype resembling Rett syndrome but with earlier onset. Most patients are unable to sit, walk, or speak. For years, <i>FOXG1</i> sequencing was only prescribed in such severe cases, limiting insight into the full clinical spectrum associated with this gene. Next-generation sequencing (NGS) now enables unbiased diagnostics. Through the European Reference Network for Rare Malformation Syndromes, Intellectual and Other Neurodevelopmental Disorders, we gathered data from patients with heterozygous <i>FOXG1</i> variants presenting a mild phenotype, defined as able to speak and walk independently. We also reviewed data from three previously reported patients meeting our criteria. We identified five new patients with pathogenic <i>FOXG1</i> missense variants, primarily in the forkhead domain, showing varying nonspecific intellectual disability and developmental delay. These features are not typical of congenital Rett syndrome and were rarely associated with microcephaly and epilepsy. Our findings are consistent with a previous genotype–phenotype analysis by Mitter et al. suggesting the delineation of five different <i>FOXG1</i> genotype groups. Milder phenotypes were associated with missense variants in the forkhead domain. This information may facilitate prognostic assessments in children carrying a <i>FOXG1</i> variant and improve the interpretation of new variants identified with genomic sequencing.</p>\",\"PeriodicalId\":7673,\"journal\":{\"name\":\"American Journal of Medical Genetics Part B: Neuropsychiatric Genetics\",\"volume\":\"195 6\",\"pages\":\"\"},\"PeriodicalIF\":1.5000,\"publicationDate\":\"2024-03-08\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/ajmg.b.32970\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"American Journal of Medical Genetics Part B: Neuropsychiatric Genetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/ajmg.b.32970\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"American Journal of Medical Genetics Part B: Neuropsychiatric Genetics","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/ajmg.b.32970","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
FOXG1 variants can be associated with milder phenotypes than congenital Rett syndrome with unassisted walking and language development
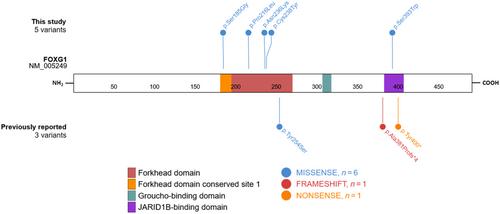
Since 2008, FOXG1 haploinsufficiency has been linked to a severe neurodevelopmental phenotype resembling Rett syndrome but with earlier onset. Most patients are unable to sit, walk, or speak. For years, FOXG1 sequencing was only prescribed in such severe cases, limiting insight into the full clinical spectrum associated with this gene. Next-generation sequencing (NGS) now enables unbiased diagnostics. Through the European Reference Network for Rare Malformation Syndromes, Intellectual and Other Neurodevelopmental Disorders, we gathered data from patients with heterozygous FOXG1 variants presenting a mild phenotype, defined as able to speak and walk independently. We also reviewed data from three previously reported patients meeting our criteria. We identified five new patients with pathogenic FOXG1 missense variants, primarily in the forkhead domain, showing varying nonspecific intellectual disability and developmental delay. These features are not typical of congenital Rett syndrome and were rarely associated with microcephaly and epilepsy. Our findings are consistent with a previous genotype–phenotype analysis by Mitter et al. suggesting the delineation of five different FOXG1 genotype groups. Milder phenotypes were associated with missense variants in the forkhead domain. This information may facilitate prognostic assessments in children carrying a FOXG1 variant and improve the interpretation of new variants identified with genomic sequencing.
期刊介绍:
Neuropsychiatric Genetics, Part B of the American Journal of Medical Genetics (AJMG) , provides a forum for experimental and clinical investigations of the genetic mechanisms underlying neurologic and psychiatric disorders. It is a resource for novel genetics studies of the heritable nature of psychiatric and other nervous system disorders, characterized at the molecular, cellular or behavior levels. Neuropsychiatric Genetics publishes eight times per year.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


