Catharina J Küng, Arezoo Daryadel, Rocio Fuente, Betül Haykir, Martin Hrabĕ de Angelis, Nati Hernando, Isabel Rubio-Aliaga, Carsten A Wagner
{"title":"一种新型家族性低钙尿症(FHH1)小鼠模型揭示了 PTH 依赖性和独立 CaSR 缺陷。","authors":"Catharina J Küng, Arezoo Daryadel, Rocio Fuente, Betül Haykir, Martin Hrabĕ de Angelis, Nati Hernando, Isabel Rubio-Aliaga, Carsten A Wagner","doi":"10.1007/s00424-024-02927-y","DOIUrl":null,"url":null,"abstract":"<p><p>The Calcium-sensing receptor (CaSR) senses extracellular calcium, regulates parathyroid hormone (PTH) secretion, and has additional functions in various organs related to systemic and local calcium and mineral homeostasis. Familial hypocalciuric hypercalcemia type I (FHH1) is caused by heterozygous loss-of-function mutations in the CaSR gene, and is characterized by the combination of hypercalcemia, hypocalciuria, normal to elevated PTH, and facultatively hypermagnesemia and mild bone mineralization defects. To date, only heterozygous Casr null mice have been available as model for FHH1. Here we present a novel mouse FHH1 model identified in a large ENU-screen that carries an c.2579 T > A (p.Ile859Asn) variant in the Casr gene (Casr<sup>BCH002</sup> mice). In order to dissect direct effects of the genetic variant from PTH-dependent effects, we crossed Casr<sup>BCH002</sup> mice with PTH deficient mice. Heterozygous Casr<sup>BCH002</sup> mice were fertile, had normal growth and body weight, were hypercalcemic and hypermagnesemic with inappropriately normal PTH levels and urinary calcium excretion replicating some features of FHH1. Hypercalcemia and hypermagnesemia were independent from PTH and correlated with higher expression of claudin 16 and 19 in kidneys. Likewise, reduced expression of the renal TRPM6 channel in Casr<sup>BCH002</sup> mice was not dependent on PTH. In bone, mutations in Casr rescued the bone phenotype observed in Pth null mice by increasing osteoclast numbers and improving the columnar pattern of chondrocytes in the growth zone. In summary, Casr<sup>BCH002</sup> mice represent a new model to study FHH1 and our results indicate that only a part of the phenotype is driven by PTH.</p>","PeriodicalId":19954,"journal":{"name":"Pflugers Archiv : European journal of physiology","volume":" ","pages":"833-845"},"PeriodicalIF":2.9000,"publicationDate":"2024-05-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11033242/pdf/","citationCount":"0","resultStr":"{\"title\":\"A novel mouse model for familial hypocalciuric hypercalcemia (FHH1) reveals PTH-dependent and independent CaSR defects.\",\"authors\":\"Catharina J Küng, Arezoo Daryadel, Rocio Fuente, Betül Haykir, Martin Hrabĕ de Angelis, Nati Hernando, Isabel Rubio-Aliaga, Carsten A Wagner\",\"doi\":\"10.1007/s00424-024-02927-y\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The Calcium-sensing receptor (CaSR) senses extracellular calcium, regulates parathyroid hormone (PTH) secretion, and has additional functions in various organs related to systemic and local calcium and mineral homeostasis. Familial hypocalciuric hypercalcemia type I (FHH1) is caused by heterozygous loss-of-function mutations in the CaSR gene, and is characterized by the combination of hypercalcemia, hypocalciuria, normal to elevated PTH, and facultatively hypermagnesemia and mild bone mineralization defects. To date, only heterozygous Casr null mice have been available as model for FHH1. Here we present a novel mouse FHH1 model identified in a large ENU-screen that carries an c.2579 T > A (p.Ile859Asn) variant in the Casr gene (Casr<sup>BCH002</sup> mice). In order to dissect direct effects of the genetic variant from PTH-dependent effects, we crossed Casr<sup>BCH002</sup> mice with PTH deficient mice. Heterozygous Casr<sup>BCH002</sup> mice were fertile, had normal growth and body weight, were hypercalcemic and hypermagnesemic with inappropriately normal PTH levels and urinary calcium excretion replicating some features of FHH1. Hypercalcemia and hypermagnesemia were independent from PTH and correlated with higher expression of claudin 16 and 19 in kidneys. Likewise, reduced expression of the renal TRPM6 channel in Casr<sup>BCH002</sup> mice was not dependent on PTH. In bone, mutations in Casr rescued the bone phenotype observed in Pth null mice by increasing osteoclast numbers and improving the columnar pattern of chondrocytes in the growth zone. In summary, Casr<sup>BCH002</sup> mice represent a new model to study FHH1 and our results indicate that only a part of the phenotype is driven by PTH.</p>\",\"PeriodicalId\":19954,\"journal\":{\"name\":\"Pflugers Archiv : European journal of physiology\",\"volume\":\" \",\"pages\":\"833-845\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2024-05-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11033242/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Pflugers Archiv : European journal of physiology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1007/s00424-024-02927-y\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/2/22 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"PHYSIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Pflugers Archiv : European journal of physiology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s00424-024-02927-y","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/2/22 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"PHYSIOLOGY","Score":null,"Total":0}
A novel mouse model for familial hypocalciuric hypercalcemia (FHH1) reveals PTH-dependent and independent CaSR defects.
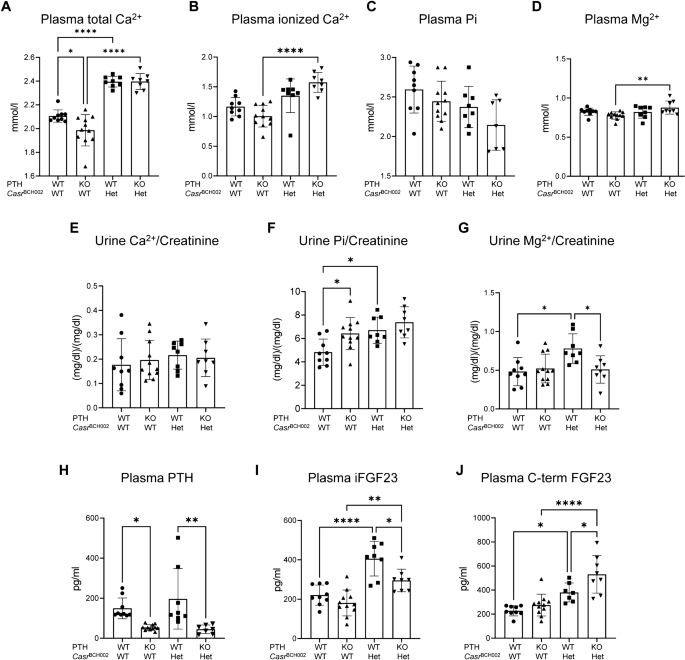
The Calcium-sensing receptor (CaSR) senses extracellular calcium, regulates parathyroid hormone (PTH) secretion, and has additional functions in various organs related to systemic and local calcium and mineral homeostasis. Familial hypocalciuric hypercalcemia type I (FHH1) is caused by heterozygous loss-of-function mutations in the CaSR gene, and is characterized by the combination of hypercalcemia, hypocalciuria, normal to elevated PTH, and facultatively hypermagnesemia and mild bone mineralization defects. To date, only heterozygous Casr null mice have been available as model for FHH1. Here we present a novel mouse FHH1 model identified in a large ENU-screen that carries an c.2579 T > A (p.Ile859Asn) variant in the Casr gene (CasrBCH002 mice). In order to dissect direct effects of the genetic variant from PTH-dependent effects, we crossed CasrBCH002 mice with PTH deficient mice. Heterozygous CasrBCH002 mice were fertile, had normal growth and body weight, were hypercalcemic and hypermagnesemic with inappropriately normal PTH levels and urinary calcium excretion replicating some features of FHH1. Hypercalcemia and hypermagnesemia were independent from PTH and correlated with higher expression of claudin 16 and 19 in kidneys. Likewise, reduced expression of the renal TRPM6 channel in CasrBCH002 mice was not dependent on PTH. In bone, mutations in Casr rescued the bone phenotype observed in Pth null mice by increasing osteoclast numbers and improving the columnar pattern of chondrocytes in the growth zone. In summary, CasrBCH002 mice represent a new model to study FHH1 and our results indicate that only a part of the phenotype is driven by PTH.
期刊介绍:
Pflügers Archiv European Journal of Physiology publishes those results of original research that are seen as advancing the physiological sciences, especially those providing mechanistic insights into physiological functions at the molecular and cellular level, and clearly conveying a physiological message. Submissions are encouraged that deal with the evaluation of molecular and cellular mechanisms of disease, ideally resulting in translational research. Purely descriptive papers covering applied physiology or clinical papers will be excluded. Papers on methodological topics will be considered if they contribute to the development of novel tools for further investigation of (patho)physiological mechanisms.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


