{"title":"通过遗传性癌症患者基因组评估目标自适应采样长读数测序的功效。","authors":"Wataru Nakamura, Makoto Hirata, Satoyo Oda, Kenichi Chiba, Ai Okada, Raúl Nicolás Mateos, Masahiro Sugawa, Naoko Iida, Mineko Ushiama, Noriko Tanabe, Hiromi Sakamoto, Shigeki Sekine, Akira Hirasawa, Yosuke Kawai, Katsushi Tokunaga, Shin-Ichi Tsujimoto, Norio Shiba, Shuichi Ito, Teruhiko Yoshida, Yuichi Shiraishi","doi":"10.1038/s41525-024-00394-z","DOIUrl":null,"url":null,"abstract":"<p><p>Innovations in sequencing technology have led to the discovery of novel mutations that cause inherited diseases. However, many patients with suspected genetic diseases remain undiagnosed. Long-read sequencing technologies are expected to significantly improve the diagnostic rate by overcoming the limitations of short-read sequencing. In addition, Oxford Nanopore Technologies (ONT) offers adaptive sampling and computationally driven target enrichment technology. This enables more affordable intensive analysis of target gene regions compared to standard non-selective long-read sequencing. In this study, we developed an efficient computational workflow for target adaptive sampling long-read sequencing (TAS-LRS) and evaluated it through application to 33 genomes collected from suspected hereditary cancer patients. Our workflow can identify single nucleotide variants with nearly the same accuracy as the short-read platform and elucidate complex forms of structural variations. We also newly identified several SINE-R/VNTR/Alu (SVA) elements affecting the APC gene in two patients with familial adenomatous polyposis, as well as their sites of origin. In addition, we demonstrated that off-target reads from adaptive sampling, which is typically discarded, can be effectively used to accurately genotype common single-nucleotide polymorphisms (SNPs) across the entire genome, enabling the calculation of a polygenic risk score. Furthermore, we identified allele-specific MLH1 promoter hypermethylation in a Lynch syndrome patient. In summary, our workflow with TAS-LRS can simultaneously capture monogenic risk variants including complex structural variations, polygenic background as well as epigenetic alterations, and will be an efficient platform for genetic disease research and diagnosis.</p>","PeriodicalId":19273,"journal":{"name":"NPJ Genomic Medicine","volume":"9 1","pages":"11"},"PeriodicalIF":4.7000,"publicationDate":"2024-02-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10874402/pdf/","citationCount":"0","resultStr":"{\"title\":\"Assessing the efficacy of target adaptive sampling long-read sequencing through hereditary cancer patient genomes.\",\"authors\":\"Wataru Nakamura, Makoto Hirata, Satoyo Oda, Kenichi Chiba, Ai Okada, Raúl Nicolás Mateos, Masahiro Sugawa, Naoko Iida, Mineko Ushiama, Noriko Tanabe, Hiromi Sakamoto, Shigeki Sekine, Akira Hirasawa, Yosuke Kawai, Katsushi Tokunaga, Shin-Ichi Tsujimoto, Norio Shiba, Shuichi Ito, Teruhiko Yoshida, Yuichi Shiraishi\",\"doi\":\"10.1038/s41525-024-00394-z\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Innovations in sequencing technology have led to the discovery of novel mutations that cause inherited diseases. However, many patients with suspected genetic diseases remain undiagnosed. Long-read sequencing technologies are expected to significantly improve the diagnostic rate by overcoming the limitations of short-read sequencing. In addition, Oxford Nanopore Technologies (ONT) offers adaptive sampling and computationally driven target enrichment technology. This enables more affordable intensive analysis of target gene regions compared to standard non-selective long-read sequencing. In this study, we developed an efficient computational workflow for target adaptive sampling long-read sequencing (TAS-LRS) and evaluated it through application to 33 genomes collected from suspected hereditary cancer patients. Our workflow can identify single nucleotide variants with nearly the same accuracy as the short-read platform and elucidate complex forms of structural variations. We also newly identified several SINE-R/VNTR/Alu (SVA) elements affecting the APC gene in two patients with familial adenomatous polyposis, as well as their sites of origin. In addition, we demonstrated that off-target reads from adaptive sampling, which is typically discarded, can be effectively used to accurately genotype common single-nucleotide polymorphisms (SNPs) across the entire genome, enabling the calculation of a polygenic risk score. Furthermore, we identified allele-specific MLH1 promoter hypermethylation in a Lynch syndrome patient. In summary, our workflow with TAS-LRS can simultaneously capture monogenic risk variants including complex structural variations, polygenic background as well as epigenetic alterations, and will be an efficient platform for genetic disease research and diagnosis.</p>\",\"PeriodicalId\":19273,\"journal\":{\"name\":\"NPJ Genomic Medicine\",\"volume\":\"9 1\",\"pages\":\"11\"},\"PeriodicalIF\":4.7000,\"publicationDate\":\"2024-02-17\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10874402/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"NPJ Genomic Medicine\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1038/s41525-024-00394-z\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"NPJ Genomic Medicine","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1038/s41525-024-00394-z","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Assessing the efficacy of target adaptive sampling long-read sequencing through hereditary cancer patient genomes.
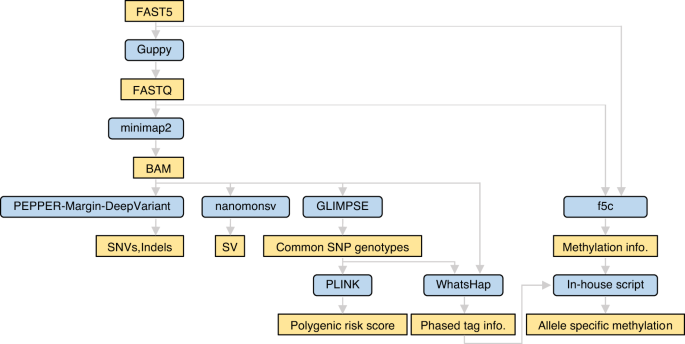
Innovations in sequencing technology have led to the discovery of novel mutations that cause inherited diseases. However, many patients with suspected genetic diseases remain undiagnosed. Long-read sequencing technologies are expected to significantly improve the diagnostic rate by overcoming the limitations of short-read sequencing. In addition, Oxford Nanopore Technologies (ONT) offers adaptive sampling and computationally driven target enrichment technology. This enables more affordable intensive analysis of target gene regions compared to standard non-selective long-read sequencing. In this study, we developed an efficient computational workflow for target adaptive sampling long-read sequencing (TAS-LRS) and evaluated it through application to 33 genomes collected from suspected hereditary cancer patients. Our workflow can identify single nucleotide variants with nearly the same accuracy as the short-read platform and elucidate complex forms of structural variations. We also newly identified several SINE-R/VNTR/Alu (SVA) elements affecting the APC gene in two patients with familial adenomatous polyposis, as well as their sites of origin. In addition, we demonstrated that off-target reads from adaptive sampling, which is typically discarded, can be effectively used to accurately genotype common single-nucleotide polymorphisms (SNPs) across the entire genome, enabling the calculation of a polygenic risk score. Furthermore, we identified allele-specific MLH1 promoter hypermethylation in a Lynch syndrome patient. In summary, our workflow with TAS-LRS can simultaneously capture monogenic risk variants including complex structural variations, polygenic background as well as epigenetic alterations, and will be an efficient platform for genetic disease research and diagnosis.
NPJ Genomic MedicineBiochemistry, Genetics and Molecular Biology-Molecular Biology
CiteScore
9.40
自引率
1.90%
发文量
67
审稿时长
17 weeks
期刊介绍:
npj Genomic Medicine is an international, peer-reviewed journal dedicated to publishing the most important scientific advances in all aspects of genomics and its application in the practice of medicine.
The journal defines genomic medicine as "diagnosis, prognosis, prevention and/or treatment of disease and disorders of the mind and body, using approaches informed or enabled by knowledge of the genome and the molecules it encodes." Relevant and high-impact papers that encompass studies of individuals, families, or populations are considered for publication. An emphasis will include coupling detailed phenotype and genome sequencing information, both enabled by new technologies and informatics, to delineate the underlying aetiology of disease. Clinical recommendations and/or guidelines of how that data should be used in the clinical management of those patients in the study, and others, are also encouraged.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


