扩展先天性肌无力综合征的遗传和表型谱:2 名新患者的新同源 VAMP1 剪接变异。
IF 2.6
3区 生物学
Q2 GENETICS & HEREDITY
引用次数: 0
摘要
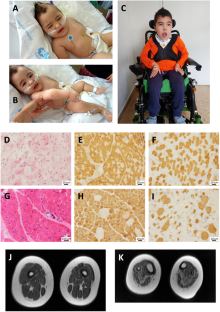
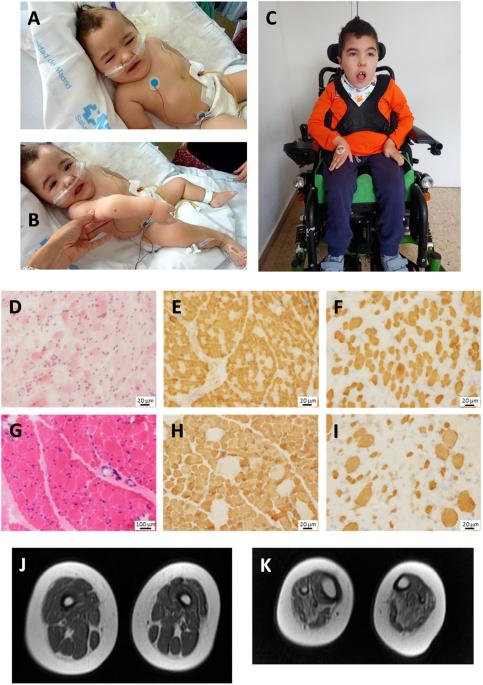
我们报告了两名西班牙儿童患者的病例,他们出生时肌张力低下、肌无力和喂养困难。全外显子组测序(WES)发现了两个新的同源 VAMP1(囊泡相关膜蛋白 1)剪接变异:男孩患者(P1)为 NM_014231.5:c.129+5 G > A,女孩患者(P2)为 c.341-24_341-16delinsAGAAAA。该基因编码囊泡相关膜蛋白 1(VAMP1),它是参与突触囊泡与突触前膜融合的蛋白质复合物的一个组成部分。VAMP1 的 C 端高度可变,通过替代剪接产生三种主要的同工型(A、B 和 D),其中 VAMP1A 是神经系统中唯一表达的同工型。为了评估这些变体的致病性,对 VAMP1 的 RNA 进行了表达实验。c.129+5 G > A和c.341-24_341-16delinsAGAAAA变体诱导了异常剪接事件,导致第2外显子缺失(r.5_131del;p.Ser2TrpfsTer7),第二种情况是在 VAMP1A 异构体中保留了内含子 4 3' 的最后 14 个核苷酸(r.340_341ins341-14_341-1;p.Ile114AsnfsTer77)。致病性 VAMP1 变异与常染色体显性痉挛性共济失调 1(SPAX1)和常染色体隐性突触前先天性肌萎缩综合征(CMS)有关。我们的患者与 CMS 患者有相同的临床表现,但有两个重要的不同点:他们没有表现出典型的电生理模式,这表明突触前神经肌肉接头出现了病变;他们的肌肉活检结果显示存在 1 型肥大纤维。总之,我们的数据扩大了与 VAMP1 变体相关的遗传和表型谱。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Expanding the genetic and phenotypic spectrum of congenital myasthenic syndrome: new homozygous VAMP1 splicing variants in 2 novel individuals
We report the cases of two Spanish pediatric patients with hypotonia, muscle weakness and feeding difficulties at birth. Whole-exome sequencing (WES) uncovered two new homozygous VAMP1 (Vesicle Associated Membrane Protein 1) splicing variants, NM_014231.5:c.129+5 G > A in the boy patient (P1) and c.341-24_341-16delinsAGAAAA in the girl patient (P2). This gene encodes the vesicle-associated membrane protein 1 (VAMP1) that is a component of a protein complex involved in the fusion of synaptic vesicles with the presynaptic membrane. VAMP1 has a highly variable C-terminus generated by alternative splicing that gives rise to three main isoforms (A, B and D), being VAMP1A the only isoform expressed in the nervous system. In order to assess the pathogenicity of these variants, expression experiments of RNA for VAMP1 were carried out. The c.129+5 G > A and c.341-24_341-16delinsAGAAAA variants induced aberrant splicing events resulting in the deletion of exon 2 (r.5_131del; p.Ser2TrpfsTer7) in the three isoforms in the first case, and the retention of the last 14 nucleotides of the 3′ of intron 4 (r.340_341ins341-14_341-1; p.Ile114AsnfsTer77) in the VAMP1A isoform in the second case. Pathogenic VAMP1 variants have been associated with autosomal dominant spastic ataxia 1 (SPAX1) and with autosomal recessive presynaptic congenital myasthenic syndrome (CMS). Our patients share the clinical manifestations of CMS patients with two important differences: they do not show the typical electrophysiological pattern that suggests pathology of pre-synaptic neuromuscular junction, and their muscular biopsies present hypertrophic fibers type 1. In conclusion, our data expand both genetic and phenotypic spectrum associated with VAMP1 variants.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Journal of Human Genetics
生物-遗传学
CiteScore
7.20
自引率
0.00%
发文量
101
审稿时长
4-8 weeks
期刊介绍:
The Journal of Human Genetics is an international journal publishing articles on human genetics, including medical genetics and human genome analysis. It covers all aspects of human genetics, including molecular genetics, clinical genetics, behavioral genetics, immunogenetics, pharmacogenomics, population genetics, functional genomics, epigenetics, genetic counseling and gene therapy.
Articles on the following areas are especially welcome: genetic factors of monogenic and complex disorders, genome-wide association studies, genetic epidemiology, cancer genetics, personal genomics, genotype-phenotype relationships and genome diversity.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


