与智力障碍、小头畸形和白内障有关的 RGS6 剪接受体变异会不成比例地促进 RGS6 同工酶亚型的表达。
IF 2.6
3区 生物学
Q2 GENETICS & HEREDITY
引用次数: 0
摘要
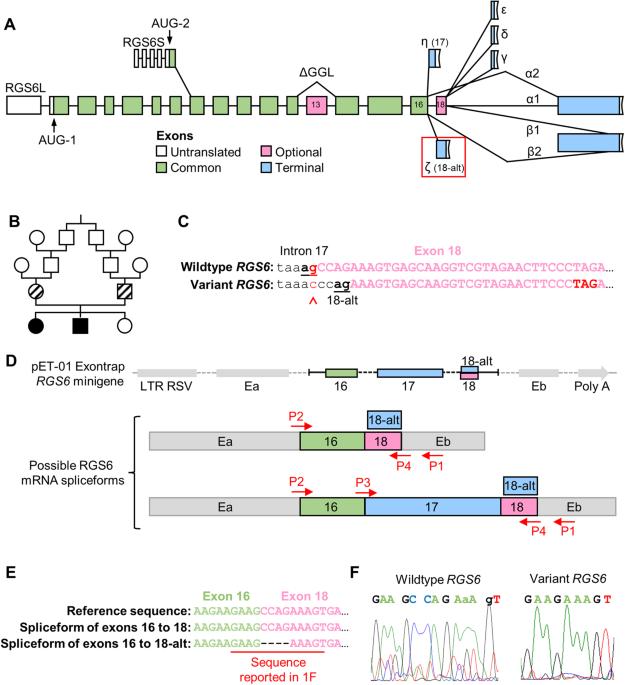
智力残疾(ID)与罹患精神疾病的风险增加有关,这表明存在共同的潜在遗传因素。重要的是,G 蛋白信号转导调节器 6(RGS6)的信号转导和/或表达改变与智障和多种精神疾病有关。RGS6 具有高度保守性,会发生复杂的 mRNA 替代剪接,产生约 36 种具有高度序列相似性的蛋白质异构体,因此有必要在功能研究中采用全局方法。然而,我们最近对小鼠的分析表明,RGS6 在中枢神经系统中的表达量最高,以 RGS6L(+GGL)异构体为主。之前报道的 RGS6 内含子 17 中的一个遗传变异(c.1369-1G>C)与 ID 有关,它可能会为 RGS6L(+GGL)异构体的功能划分提供进一步的线索。据预测,该变异会改变一个高度保守的 3' 接受位点,在第 18 号外显子(包含在 RGS6L(+GGL) 转录本的一个子集中)内产生一个替代分支点,并形成一个早期终止密码子的框架移位。我们以前曾鉴定过这个替代剪接位点,并证明利用它可以产生 RGS6Lζ(+GGL) 异构体。在这里,我们发现 c.1369-1G>C 变体破坏了规范的、首选的(>90%)第 17 号内含子剪接位点,导致只能使用第 18 号外显子的替代剪接位点,从而诱导了一部分同工酶,特别是 RGS6Lζ(+GGL) 的过度表达。此外,RGS6 基因全基因敲除小鼠不表现出 ID。因此,c.1369-1G>C 变异导致的 ID 很可能是 RGS6 同工酶表达的改变,而不是 RGS6 同工酶的缺失。总之,这些研究强调了正确的 RGS6 剪接的重要性,并确定了 G 蛋白信号在 ID 中以前未被认识到的作用。本文章由计算机程序翻译,如有差异,请以英文原文为准。
A splice acceptor variant in RGS6 associated with intellectual disability, microcephaly, and cataracts disproportionately promotes expression of a subset of RGS6 isoforms
Intellectual disability (ID) is associated with an increased risk of developing psychiatric disorders, suggesting a common underlying genetic factor. Importantly, altered signaling and/or expression of regulator of G protein signaling 6 (RGS6) is associated with ID and numerous psychiatric disorders. RGS6 is highly conserved and undergoes complex alternative mRNA splicing producing ~36 protein isoforms with high sequence similarity historically necessitating a global approach in functional studies. However, our recent analysis in mice revealed RGS6 is most highly expressed in CNS with RGS6L(+GGL) isoforms predominating. A previously reported genetic variant in intron 17 of RGS6 (c.1369-1G>C), associated with ID, may provide further clues into RGS6L(+GGL) isoform functional delineation. This variant was predicted to alter a highly conserved canonical 3’ acceptor site creating an alternative branch point within exon 18 (included in a subset of RGS6L(+GGL) transcripts) and a frameshift forming an early stop codon. We previously identified this alternative splice site and demonstrated its use generates RGS6Lζ(+GGL) isoforms. Here, we show that the c.1369-1G>C variant disrupts the canonical, preferred (>90%) intron 17 splice site and leads to the exclusive use of the alternate exon 18 splice site, inducing disproportionate expression of a subset of isoforms, particularly RGS6Lζ(+GGL). Furthermore, RGS6 global knockout mice do not exhibit ID. Thus, ID caused by the c.1369-1G>C variant likely results from altered RGS6 isoform expression, rather than RGS6 isoform loss. In summary, these studies highlight the importance of proper RGS6 splicing and identify a previously unrecognized role of G protein signaling in ID.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Journal of Human Genetics
生物-遗传学
CiteScore
7.20
自引率
0.00%
发文量
101
审稿时长
4-8 weeks
期刊介绍:
The Journal of Human Genetics is an international journal publishing articles on human genetics, including medical genetics and human genome analysis. It covers all aspects of human genetics, including molecular genetics, clinical genetics, behavioral genetics, immunogenetics, pharmacogenomics, population genetics, functional genomics, epigenetics, genetic counseling and gene therapy.
Articles on the following areas are especially welcome: genetic factors of monogenic and complex disorders, genome-wide association studies, genetic epidemiology, cancer genetics, personal genomics, genotype-phenotype relationships and genome diversity.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


