Claude Bhérer, Robert Eveleigh, Katerina Trajanoska, Janick St-Cyr, Antoine Paccard, Praveen Nadukkalam Ravindran, Elizabeth Caron, Nimara Bader Asbah, Peyton McClelland, Clare Wei, Iris Baumgartner, Marc Schindewolf, Yvonne Döring, Danielle Perley, François Lefebvre, Pierre Lepage, Mathieu Bourgey, Guillaume Bourque, Jiannis Ragoussis, Vincent Mooser, Daniel Taliun
{"title":"一种结合高深度全外显子组和低深度全基因组的经济高效的基因研究测序方法。","authors":"Claude Bhérer, Robert Eveleigh, Katerina Trajanoska, Janick St-Cyr, Antoine Paccard, Praveen Nadukkalam Ravindran, Elizabeth Caron, Nimara Bader Asbah, Peyton McClelland, Clare Wei, Iris Baumgartner, Marc Schindewolf, Yvonne Döring, Danielle Perley, François Lefebvre, Pierre Lepage, Mathieu Bourgey, Guillaume Bourque, Jiannis Ragoussis, Vincent Mooser, Daniel Taliun","doi":"10.1038/s41525-024-00390-3","DOIUrl":null,"url":null,"abstract":"<p><p>Whole genome sequencing (WGS) at high-depth (30X) allows the accurate discovery of variants in the coding and non-coding DNA regions and helps elucidate the genetic underpinnings of human health and diseases. Yet, due to the prohibitive cost of high-depth WGS, most large-scale genetic association studies use genotyping arrays or high-depth whole exome sequencing (WES). Here we propose a cost-effective method which we call \"Whole Exome Genome Sequencing\" (WEGS), that combines low-depth WGS and high-depth WES with up to 8 samples pooled and sequenced simultaneously (multiplexed). We experimentally assess the performance of WEGS with four different depth of coverage and sample multiplexing configurations. We show that the optimal WEGS configurations are 1.7-2.0 times cheaper than standard WES (no-plexing), 1.8-2.1 times cheaper than high-depth WGS, reach similar recall and precision rates in detecting coding variants as WES, and capture more population-specific variants in the rest of the genome that are difficult to recover when using genotype imputation methods. We apply WEGS to 862 patients with peripheral artery disease and show that it directly assesses more known disease-associated variants than a typical genotyping array and thousands of non-imputable variants per disease-associated locus.</p>","PeriodicalId":19273,"journal":{"name":"NPJ Genomic Medicine","volume":"9 1","pages":"8"},"PeriodicalIF":4.7000,"publicationDate":"2024-02-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10850497/pdf/","citationCount":"0","resultStr":"{\"title\":\"A cost-effective sequencing method for genetic studies combining high-depth whole exome and low-depth whole genome.\",\"authors\":\"Claude Bhérer, Robert Eveleigh, Katerina Trajanoska, Janick St-Cyr, Antoine Paccard, Praveen Nadukkalam Ravindran, Elizabeth Caron, Nimara Bader Asbah, Peyton McClelland, Clare Wei, Iris Baumgartner, Marc Schindewolf, Yvonne Döring, Danielle Perley, François Lefebvre, Pierre Lepage, Mathieu Bourgey, Guillaume Bourque, Jiannis Ragoussis, Vincent Mooser, Daniel Taliun\",\"doi\":\"10.1038/s41525-024-00390-3\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Whole genome sequencing (WGS) at high-depth (30X) allows the accurate discovery of variants in the coding and non-coding DNA regions and helps elucidate the genetic underpinnings of human health and diseases. Yet, due to the prohibitive cost of high-depth WGS, most large-scale genetic association studies use genotyping arrays or high-depth whole exome sequencing (WES). Here we propose a cost-effective method which we call \\\"Whole Exome Genome Sequencing\\\" (WEGS), that combines low-depth WGS and high-depth WES with up to 8 samples pooled and sequenced simultaneously (multiplexed). We experimentally assess the performance of WEGS with four different depth of coverage and sample multiplexing configurations. We show that the optimal WEGS configurations are 1.7-2.0 times cheaper than standard WES (no-plexing), 1.8-2.1 times cheaper than high-depth WGS, reach similar recall and precision rates in detecting coding variants as WES, and capture more population-specific variants in the rest of the genome that are difficult to recover when using genotype imputation methods. We apply WEGS to 862 patients with peripheral artery disease and show that it directly assesses more known disease-associated variants than a typical genotyping array and thousands of non-imputable variants per disease-associated locus.</p>\",\"PeriodicalId\":19273,\"journal\":{\"name\":\"NPJ Genomic Medicine\",\"volume\":\"9 1\",\"pages\":\"8\"},\"PeriodicalIF\":4.7000,\"publicationDate\":\"2024-02-07\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10850497/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"NPJ Genomic Medicine\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1038/s41525-024-00390-3\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"NPJ Genomic Medicine","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1038/s41525-024-00390-3","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
A cost-effective sequencing method for genetic studies combining high-depth whole exome and low-depth whole genome.
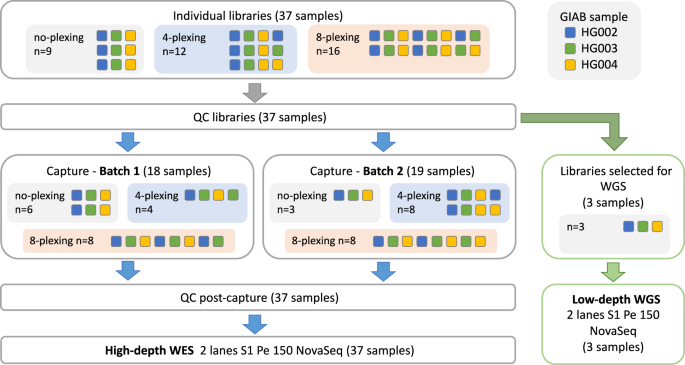
Whole genome sequencing (WGS) at high-depth (30X) allows the accurate discovery of variants in the coding and non-coding DNA regions and helps elucidate the genetic underpinnings of human health and diseases. Yet, due to the prohibitive cost of high-depth WGS, most large-scale genetic association studies use genotyping arrays or high-depth whole exome sequencing (WES). Here we propose a cost-effective method which we call "Whole Exome Genome Sequencing" (WEGS), that combines low-depth WGS and high-depth WES with up to 8 samples pooled and sequenced simultaneously (multiplexed). We experimentally assess the performance of WEGS with four different depth of coverage and sample multiplexing configurations. We show that the optimal WEGS configurations are 1.7-2.0 times cheaper than standard WES (no-plexing), 1.8-2.1 times cheaper than high-depth WGS, reach similar recall and precision rates in detecting coding variants as WES, and capture more population-specific variants in the rest of the genome that are difficult to recover when using genotype imputation methods. We apply WEGS to 862 patients with peripheral artery disease and show that it directly assesses more known disease-associated variants than a typical genotyping array and thousands of non-imputable variants per disease-associated locus.
NPJ Genomic MedicineBiochemistry, Genetics and Molecular Biology-Molecular Biology
CiteScore
9.40
自引率
1.90%
发文量
67
审稿时长
17 weeks
期刊介绍:
npj Genomic Medicine is an international, peer-reviewed journal dedicated to publishing the most important scientific advances in all aspects of genomics and its application in the practice of medicine.
The journal defines genomic medicine as "diagnosis, prognosis, prevention and/or treatment of disease and disorders of the mind and body, using approaches informed or enabled by knowledge of the genome and the molecules it encodes." Relevant and high-impact papers that encompass studies of individuals, families, or populations are considered for publication. An emphasis will include coupling detailed phenotype and genome sequencing information, both enabled by new technologies and informatics, to delineate the underlying aetiology of disease. Clinical recommendations and/or guidelines of how that data should be used in the clinical management of those patients in the study, and others, are also encouraged.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


