Divya C. Ragate, Saba Samad Memon, Manjiri Karlekar, Anurag Ranjan Lila, Vijaya Sarathi, Tukaram Jamale, Sayali Thakare, Virendra A. Patil, Nalini S. Shah, Tushar R. Bandgar
{"title":"遗传性范可尼肾小管综合征:揭开低磷血症佝偻病/骨软化症的神秘面纱","authors":"Divya C. Ragate, Saba Samad Memon, Manjiri Karlekar, Anurag Ranjan Lila, Vijaya Sarathi, Tukaram Jamale, Sayali Thakare, Virendra A. Patil, Nalini S. Shah, Tushar R. Bandgar","doi":"10.1007/s00774-023-01490-3","DOIUrl":null,"url":null,"abstract":"<h3 data-test=\"abstract-sub-heading\">Introduction</h3><p>Fanconi renotubular syndromes (FRTS) are a rare group of inherited phosphaturic disorders with limited Indian as well as global data on this condition. Here, we describe the experience of a single Endocrinology center from Western India on FRTS.</p><h3 data-test=\"abstract-sub-heading\">Materials and methods</h3><p>Comprehensive clinical, biochemical, radiological, management, and genetic details of FRTS patients managed between 2010 and 2023 were collected and analyzed.</p><h3 data-test=\"abstract-sub-heading\">Results</h3><p>FRTS probands had mutations (eight novel) in six genes [<i>CLCN5</i> (<i>n</i> = 4)<i>, SLC2A2</i> (<i>n</i> = 2<i>), GATM</i>, <i>EHHADH, HNF4A,</i> and <i>OCRL </i>(1 each)]. Among 15 FRTS patients (11 families), rickets/osteomalacia was the most common (<i>n</i> = 14) presentation with wide inter- and intra-familial phenotypic variability. Delayed diagnosis (median: 8.8 years), initial misdiagnosis (8/11 probands), and syndrome-specific discriminatory features (8/11 probands) were commonly seen. Hypophosphatemia, elevated alkaline phosphatase, normal parathyroid hormone (median: 36 pg/ml), high-normal/elevated 1,25(OH)<sub>2</sub>D (median: 152 pg/ml), hypercalciuria (median spot urinary calcium to creatinine ratio: 0.32), and variable proximal tubular dysfunction(s) were observed. Elevated C-terminal fibroblast growth factor 23 in two probands was misleading, till the genetic diagnosis was reached. Novel observations in our FRTS cohort were preserved renal function (till sixth decade) and enthesopathy in FRTS1 and FRTS3 families, respectively.</p><h3 data-test=\"abstract-sub-heading\">Conclusion</h3><p>Our findings underscore frequent under- and misdiagnosis of FRTS; hence, a high index of suspicion for FRTS in phosphopenic rickets/osteomalacia, with early consideration of genetic testing is essential to ensure timely diagnosis of FRTS. The novel variants and phenotypic manifestations described here expand the disease spectrum of FRTS.</p>","PeriodicalId":15116,"journal":{"name":"Journal of Bone and Mineral Metabolism","volume":"254 1","pages":""},"PeriodicalIF":2.4000,"publicationDate":"2024-02-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Inherited Fanconi renotubular syndromes: unveiling the intricacies of hypophosphatemic rickets/osteomalacia\",\"authors\":\"Divya C. Ragate, Saba Samad Memon, Manjiri Karlekar, Anurag Ranjan Lila, Vijaya Sarathi, Tukaram Jamale, Sayali Thakare, Virendra A. Patil, Nalini S. Shah, Tushar R. Bandgar\",\"doi\":\"10.1007/s00774-023-01490-3\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<h3 data-test=\\\"abstract-sub-heading\\\">Introduction</h3><p>Fanconi renotubular syndromes (FRTS) are a rare group of inherited phosphaturic disorders with limited Indian as well as global data on this condition. Here, we describe the experience of a single Endocrinology center from Western India on FRTS.</p><h3 data-test=\\\"abstract-sub-heading\\\">Materials and methods</h3><p>Comprehensive clinical, biochemical, radiological, management, and genetic details of FRTS patients managed between 2010 and 2023 were collected and analyzed.</p><h3 data-test=\\\"abstract-sub-heading\\\">Results</h3><p>FRTS probands had mutations (eight novel) in six genes [<i>CLCN5</i> (<i>n</i> = 4)<i>, SLC2A2</i> (<i>n</i> = 2<i>), GATM</i>, <i>EHHADH, HNF4A,</i> and <i>OCRL </i>(1 each)]. Among 15 FRTS patients (11 families), rickets/osteomalacia was the most common (<i>n</i> = 14) presentation with wide inter- and intra-familial phenotypic variability. Delayed diagnosis (median: 8.8 years), initial misdiagnosis (8/11 probands), and syndrome-specific discriminatory features (8/11 probands) were commonly seen. Hypophosphatemia, elevated alkaline phosphatase, normal parathyroid hormone (median: 36 pg/ml), high-normal/elevated 1,25(OH)<sub>2</sub>D (median: 152 pg/ml), hypercalciuria (median spot urinary calcium to creatinine ratio: 0.32), and variable proximal tubular dysfunction(s) were observed. Elevated C-terminal fibroblast growth factor 23 in two probands was misleading, till the genetic diagnosis was reached. Novel observations in our FRTS cohort were preserved renal function (till sixth decade) and enthesopathy in FRTS1 and FRTS3 families, respectively.</p><h3 data-test=\\\"abstract-sub-heading\\\">Conclusion</h3><p>Our findings underscore frequent under- and misdiagnosis of FRTS; hence, a high index of suspicion for FRTS in phosphopenic rickets/osteomalacia, with early consideration of genetic testing is essential to ensure timely diagnosis of FRTS. The novel variants and phenotypic manifestations described here expand the disease spectrum of FRTS.</p>\",\"PeriodicalId\":15116,\"journal\":{\"name\":\"Journal of Bone and Mineral Metabolism\",\"volume\":\"254 1\",\"pages\":\"\"},\"PeriodicalIF\":2.4000,\"publicationDate\":\"2024-02-04\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Bone and Mineral Metabolism\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1007/s00774-023-01490-3\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"ENDOCRINOLOGY & METABOLISM\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Bone and Mineral Metabolism","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s00774-023-01490-3","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
Inherited Fanconi renotubular syndromes: unveiling the intricacies of hypophosphatemic rickets/osteomalacia
Introduction
Fanconi renotubular syndromes (FRTS) are a rare group of inherited phosphaturic disorders with limited Indian as well as global data on this condition. Here, we describe the experience of a single Endocrinology center from Western India on FRTS.
Materials and methods
Comprehensive clinical, biochemical, radiological, management, and genetic details of FRTS patients managed between 2010 and 2023 were collected and analyzed.
Results
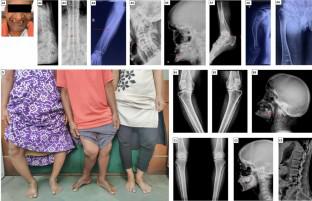
FRTS probands had mutations (eight novel) in six genes [CLCN5 (n = 4), SLC2A2 (n = 2), GATM, EHHADH, HNF4A, and OCRL (1 each)]. Among 15 FRTS patients (11 families), rickets/osteomalacia was the most common (n = 14) presentation with wide inter- and intra-familial phenotypic variability. Delayed diagnosis (median: 8.8 years), initial misdiagnosis (8/11 probands), and syndrome-specific discriminatory features (8/11 probands) were commonly seen. Hypophosphatemia, elevated alkaline phosphatase, normal parathyroid hormone (median: 36 pg/ml), high-normal/elevated 1,25(OH)2D (median: 152 pg/ml), hypercalciuria (median spot urinary calcium to creatinine ratio: 0.32), and variable proximal tubular dysfunction(s) were observed. Elevated C-terminal fibroblast growth factor 23 in two probands was misleading, till the genetic diagnosis was reached. Novel observations in our FRTS cohort were preserved renal function (till sixth decade) and enthesopathy in FRTS1 and FRTS3 families, respectively.
Conclusion
Our findings underscore frequent under- and misdiagnosis of FRTS; hence, a high index of suspicion for FRTS in phosphopenic rickets/osteomalacia, with early consideration of genetic testing is essential to ensure timely diagnosis of FRTS. The novel variants and phenotypic manifestations described here expand the disease spectrum of FRTS.
期刊介绍:
The Journal of Bone and Mineral Metabolism (JBMM) provides an international forum for researchers and clinicians to present and discuss topics relevant to bone, teeth, and mineral metabolism, as well as joint and musculoskeletal disorders. The journal welcomes the submission of manuscripts from any country. Membership in the society is not a prerequisite for submission. Acceptance is based on the originality, significance, and validity of the material presented. The journal is aimed at researchers and clinicians dedicated to improvements in research, development, and patient-care in the fields of bone and mineral metabolism.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


