Klemens Fröhlich, Regula Furrer, Christian Schori, Christoph Handschin and Alexander Schmidt*,
{"title":"使用小质量范围和窄窗口数据独立采集方案进行稳健、精确和深入的蛋白质组分析。","authors":"Klemens Fröhlich, Regula Furrer, Christian Schori, Christoph Handschin and Alexander Schmidt*, ","doi":"10.1021/acs.jproteome.3c00736","DOIUrl":null,"url":null,"abstract":"<p >In recent years, a plethora of different data-independent acquisition methods have been developed for proteomics to cover a wide range of requirements. Current deep proteome profiling methods rely on fractionations, elaborate chromatography, and mass spectrometry setups or display suboptimal quantitative precision. We set out to develop an easy-to-use one shot DIA method that achieves high quantitative precision and high proteome coverage. We achieve this by focusing on a small mass range of 430–670 <i>m</i>/<i>z</i> using small isolation windows without overlap. With this new method, we were able to quantify >9200 protein groups in HEK lysates with an average coefficient of variance of 3.2%. To demonstrate the power of our newly developed narrow mass range method, we applied it to investigate the effect of PGC-1α knockout on the skeletal muscle proteome in mice. Compared to a standard data-dependent acquisition method, we could double proteome coverage and, most importantly, achieve a significantly higher quantitative precision, as compared to a previously proposed DIA method. We believe that our method will be especially helpful in quantifying low abundant proteins in samples with a high dynamic range. All raw and result files are available at massive.ucsd.edu (MSV000092186).</p>","PeriodicalId":48,"journal":{"name":"Journal of Proteome Research","volume":"23 3","pages":"1028–1038"},"PeriodicalIF":3.6000,"publicationDate":"2024-01-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.acs.org/doi/epdf/10.1021/acs.jproteome.3c00736","citationCount":"0","resultStr":"{\"title\":\"Robust, Precise, and Deep Proteome Profiling Using a Small Mass Range and Narrow Window Data-Independent-Acquisition Scheme\",\"authors\":\"Klemens Fröhlich, Regula Furrer, Christian Schori, Christoph Handschin and Alexander Schmidt*, \",\"doi\":\"10.1021/acs.jproteome.3c00736\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >In recent years, a plethora of different data-independent acquisition methods have been developed for proteomics to cover a wide range of requirements. Current deep proteome profiling methods rely on fractionations, elaborate chromatography, and mass spectrometry setups or display suboptimal quantitative precision. We set out to develop an easy-to-use one shot DIA method that achieves high quantitative precision and high proteome coverage. We achieve this by focusing on a small mass range of 430–670 <i>m</i>/<i>z</i> using small isolation windows without overlap. With this new method, we were able to quantify >9200 protein groups in HEK lysates with an average coefficient of variance of 3.2%. To demonstrate the power of our newly developed narrow mass range method, we applied it to investigate the effect of PGC-1α knockout on the skeletal muscle proteome in mice. Compared to a standard data-dependent acquisition method, we could double proteome coverage and, most importantly, achieve a significantly higher quantitative precision, as compared to a previously proposed DIA method. We believe that our method will be especially helpful in quantifying low abundant proteins in samples with a high dynamic range. All raw and result files are available at massive.ucsd.edu (MSV000092186).</p>\",\"PeriodicalId\":48,\"journal\":{\"name\":\"Journal of Proteome Research\",\"volume\":\"23 3\",\"pages\":\"1028–1038\"},\"PeriodicalIF\":3.6000,\"publicationDate\":\"2024-01-26\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.acs.org/doi/epdf/10.1021/acs.jproteome.3c00736\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Proteome Research\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jproteome.3c00736\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Proteome Research","FirstCategoryId":"99","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jproteome.3c00736","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
摘要
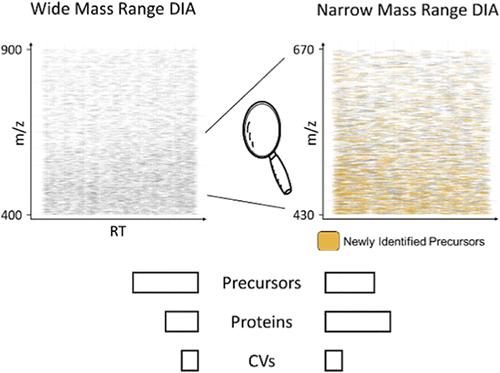
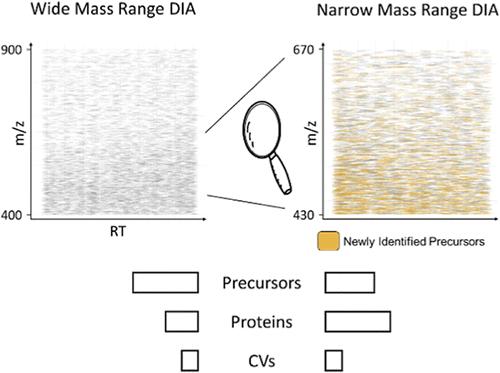
近年来,针对蛋白质组学开发了大量不同的独立于数据的采集方法,以满足广泛的要求。目前的深度蛋白质组分析方法依赖于分馏、复杂的色谱和质谱设置,或显示出不理想的定量精度。我们的目标是开发一种易于使用的一次性 DIA 方法,实现高定量精度和高蛋白质组覆盖率。我们通过使用无重叠的小分离窗口,聚焦于 430-670 m/z 的小质量范围来实现这一目标。利用这种新方法,我们能够定量检测 HEK 裂解物中超过 9200 个蛋白质组,平均方差系数为 3.2%。为了证明我们新开发的窄质量范围方法的能力,我们将其用于研究 PGC-1α 基因敲除对小鼠骨骼肌蛋白质组的影响。与标准的数据依赖性采集方法相比,我们的蛋白质组覆盖率提高了一倍,更重要的是,与之前提出的 DIA 方法相比,我们的定量精度显著提高。我们相信,我们的方法尤其有助于定量高动态范围样本中含量较低的蛋白质。所有原始文件和结果文件均可在 massive.ucsd.edu (MSV000092186) 上查阅。
Robust, Precise, and Deep Proteome Profiling Using a Small Mass Range and Narrow Window Data-Independent-Acquisition Scheme
In recent years, a plethora of different data-independent acquisition methods have been developed for proteomics to cover a wide range of requirements. Current deep proteome profiling methods rely on fractionations, elaborate chromatography, and mass spectrometry setups or display suboptimal quantitative precision. We set out to develop an easy-to-use one shot DIA method that achieves high quantitative precision and high proteome coverage. We achieve this by focusing on a small mass range of 430–670 m/z using small isolation windows without overlap. With this new method, we were able to quantify >9200 protein groups in HEK lysates with an average coefficient of variance of 3.2%. To demonstrate the power of our newly developed narrow mass range method, we applied it to investigate the effect of PGC-1α knockout on the skeletal muscle proteome in mice. Compared to a standard data-dependent acquisition method, we could double proteome coverage and, most importantly, achieve a significantly higher quantitative precision, as compared to a previously proposed DIA method. We believe that our method will be especially helpful in quantifying low abundant proteins in samples with a high dynamic range. All raw and result files are available at massive.ucsd.edu (MSV000092186).
期刊介绍:
Journal of Proteome Research publishes content encompassing all aspects of global protein analysis and function, including the dynamic aspects of genomics, spatio-temporal proteomics, metabonomics and metabolomics, clinical and agricultural proteomics, as well as advances in methodology including bioinformatics. The theme and emphasis is on a multidisciplinary approach to the life sciences through the synergy between the different types of "omics".

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


