Hanna G Budayeva, Taylur P Ma, Shuai Wang, Meena Choi, Christopher M Rose
{"title":"利用超复用和智能数据采集提高基于活动的蛋白质组轮廓分析研究的通量和可重复性。","authors":"Hanna G Budayeva, Taylur P Ma, Shuai Wang, Meena Choi, Christopher M Rose","doi":"10.1021/acs.jproteome.3c00598","DOIUrl":null,"url":null,"abstract":"<p><p>Intelligent data acquisition (IDA) strategies, such as a real-time database search (RTS), have improved the depth of proteome coverage for experiments that utilize isobaric labels and gas phase purification techniques (i.e., SPS-MS3). In this work, we introduce inSeqAPI, an instrument application programing interface (iAPI) program that enables construction of novel data acquisition algorithms. First, we analyze biotinylated cysteine peptides from ABPP experiments to demonstrate that a real-time search method within inSeqAPI performs similarly to an equivalent vendor method. Then, we describe PairQuant, a method within inSeqAPI designed for the hyperplexing approach that utilizes protein-level isotopic labeling and peptide-level TMT labeling. PairQuant allows for TMT analysis of 36 conditions in a single sample and achieves ∼98% coverage of both peptide pair partners in a hyperplexed experiment as well as a 40% improvement in the number of quantified cysteine sites compared with non-RTS acquisition. We applied this method in the ABPP study of ligandable cysteine sites in the nucleus leading to an identification of additional druggable sites on protein- and DNA-interaction domains of transcription regulators and on nuclear ubiquitin ligases.</p>","PeriodicalId":48,"journal":{"name":"Journal of Proteome Research","volume":" ","pages":"2934-2947"},"PeriodicalIF":3.6000,"publicationDate":"2024-08-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11301772/pdf/","citationCount":"0","resultStr":"{\"title\":\"Increasing the Throughput and Reproducibility of Activity-Based Proteome Profiling Studies with Hyperplexing and Intelligent Data Acquisition.\",\"authors\":\"Hanna G Budayeva, Taylur P Ma, Shuai Wang, Meena Choi, Christopher M Rose\",\"doi\":\"10.1021/acs.jproteome.3c00598\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Intelligent data acquisition (IDA) strategies, such as a real-time database search (RTS), have improved the depth of proteome coverage for experiments that utilize isobaric labels and gas phase purification techniques (i.e., SPS-MS3). In this work, we introduce inSeqAPI, an instrument application programing interface (iAPI) program that enables construction of novel data acquisition algorithms. First, we analyze biotinylated cysteine peptides from ABPP experiments to demonstrate that a real-time search method within inSeqAPI performs similarly to an equivalent vendor method. Then, we describe PairQuant, a method within inSeqAPI designed for the hyperplexing approach that utilizes protein-level isotopic labeling and peptide-level TMT labeling. PairQuant allows for TMT analysis of 36 conditions in a single sample and achieves ∼98% coverage of both peptide pair partners in a hyperplexed experiment as well as a 40% improvement in the number of quantified cysteine sites compared with non-RTS acquisition. We applied this method in the ABPP study of ligandable cysteine sites in the nucleus leading to an identification of additional druggable sites on protein- and DNA-interaction domains of transcription regulators and on nuclear ubiquitin ligases.</p>\",\"PeriodicalId\":48,\"journal\":{\"name\":\"Journal of Proteome Research\",\"volume\":\" \",\"pages\":\"2934-2947\"},\"PeriodicalIF\":3.6000,\"publicationDate\":\"2024-08-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11301772/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Proteome Research\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jproteome.3c00598\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/1/22 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Proteome Research","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1021/acs.jproteome.3c00598","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/22 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
Increasing the Throughput and Reproducibility of Activity-Based Proteome Profiling Studies with Hyperplexing and Intelligent Data Acquisition.
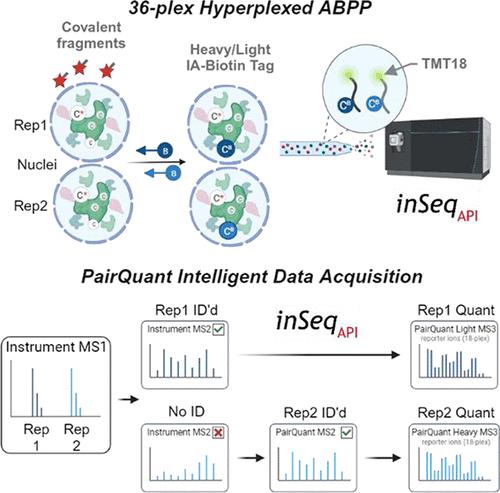
Intelligent data acquisition (IDA) strategies, such as a real-time database search (RTS), have improved the depth of proteome coverage for experiments that utilize isobaric labels and gas phase purification techniques (i.e., SPS-MS3). In this work, we introduce inSeqAPI, an instrument application programing interface (iAPI) program that enables construction of novel data acquisition algorithms. First, we analyze biotinylated cysteine peptides from ABPP experiments to demonstrate that a real-time search method within inSeqAPI performs similarly to an equivalent vendor method. Then, we describe PairQuant, a method within inSeqAPI designed for the hyperplexing approach that utilizes protein-level isotopic labeling and peptide-level TMT labeling. PairQuant allows for TMT analysis of 36 conditions in a single sample and achieves ∼98% coverage of both peptide pair partners in a hyperplexed experiment as well as a 40% improvement in the number of quantified cysteine sites compared with non-RTS acquisition. We applied this method in the ABPP study of ligandable cysteine sites in the nucleus leading to an identification of additional druggable sites on protein- and DNA-interaction domains of transcription regulators and on nuclear ubiquitin ligases.
期刊介绍:
Journal of Proteome Research publishes content encompassing all aspects of global protein analysis and function, including the dynamic aspects of genomics, spatio-temporal proteomics, metabonomics and metabolomics, clinical and agricultural proteomics, as well as advances in methodology including bioinformatics. The theme and emphasis is on a multidisciplinary approach to the life sciences through the synergy between the different types of "omics".

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


