{"title":"量化氧化石墨烯结构中的缺陷","authors":"Sownyak Mondal, Soumya Ghosh","doi":"10.1016/j.cartre.2024.100323","DOIUrl":null,"url":null,"abstract":"<div><p>Oxidation of graphite and subsequent exfoliation leads to single layer graphene oxide (GO). One of the key structural features of GO is the presence of different kinds of defects that dictates their various physical and chemical properties. Molecular dynamics simulations with ReaxFF force fields have been widely used to generate realistic models of GO. In these simulations, the extent and distribution of the defects are varied by changing the initial oxygen (O)/carbon (C) ratio while the defect density is often measured by the total number of non-graphitic carbon (non-gC) atoms. Our calculations suggest that this parameter overestimates the defect densities at low O/C ratio. Herein, we employ the relative area of the defects as an alternative metric to gauge the defect density. Being exclusive to the defects and sensitive to the structural irregularities, this metric works well at both low and high defect densities. In another example, we consider the reduction of GO to reduced graphene oxide (rGO) for different O/C ratios, where the decrease in the number of non-gC atoms is associated with the formation of comparatively larger defects. Hence, it is unclear whether the extent of reduction in the defect density (if at all) should vary monotonically with the O/C ratio. The defect area, unlike the count of the non-gC atoms, mirrors this ambiguity and the change with respect to O/C ratio is not strictly monotonic. Additionally, we also investigate the dependence of the defect distribution and defect area on the size of the simulation cell.</p></div>","PeriodicalId":52629,"journal":{"name":"Carbon Trends","volume":"14 ","pages":"Article 100323"},"PeriodicalIF":3.1000,"publicationDate":"2024-01-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S266705692400004X/pdfft?md5=dcf53da0c17aa7fddc50bcf02631ec93&pid=1-s2.0-S266705692400004X-main.pdf","citationCount":"0","resultStr":"{\"title\":\"Quantifying defects in graphene oxide structures\",\"authors\":\"Sownyak Mondal, Soumya Ghosh\",\"doi\":\"10.1016/j.cartre.2024.100323\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Oxidation of graphite and subsequent exfoliation leads to single layer graphene oxide (GO). One of the key structural features of GO is the presence of different kinds of defects that dictates their various physical and chemical properties. Molecular dynamics simulations with ReaxFF force fields have been widely used to generate realistic models of GO. In these simulations, the extent and distribution of the defects are varied by changing the initial oxygen (O)/carbon (C) ratio while the defect density is often measured by the total number of non-graphitic carbon (non-gC) atoms. Our calculations suggest that this parameter overestimates the defect densities at low O/C ratio. Herein, we employ the relative area of the defects as an alternative metric to gauge the defect density. Being exclusive to the defects and sensitive to the structural irregularities, this metric works well at both low and high defect densities. In another example, we consider the reduction of GO to reduced graphene oxide (rGO) for different O/C ratios, where the decrease in the number of non-gC atoms is associated with the formation of comparatively larger defects. Hence, it is unclear whether the extent of reduction in the defect density (if at all) should vary monotonically with the O/C ratio. The defect area, unlike the count of the non-gC atoms, mirrors this ambiguity and the change with respect to O/C ratio is not strictly monotonic. Additionally, we also investigate the dependence of the defect distribution and defect area on the size of the simulation cell.</p></div>\",\"PeriodicalId\":52629,\"journal\":{\"name\":\"Carbon Trends\",\"volume\":\"14 \",\"pages\":\"Article 100323\"},\"PeriodicalIF\":3.1000,\"publicationDate\":\"2024-01-12\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.sciencedirect.com/science/article/pii/S266705692400004X/pdfft?md5=dcf53da0c17aa7fddc50bcf02631ec93&pid=1-s2.0-S266705692400004X-main.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Carbon Trends\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S266705692400004X\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"MATERIALS SCIENCE, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Carbon Trends","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S266705692400004X","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"MATERIALS SCIENCE, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
摘要
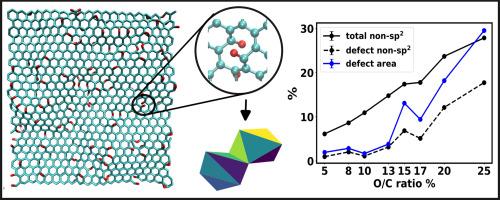
石墨氧化和随后的剥离会产生单层氧化石墨烯(GO)。GO 的主要结构特征之一是存在不同种类的缺陷,这些缺陷决定了其各种物理和化学特性。利用 ReaxFF 力场进行的分子动力学模拟已被广泛用于生成逼真的 GO 模型。在这些模拟中,缺陷的程度和分布是通过改变初始氧(O)/碳(C)比率来改变的,而缺陷密度通常是通过非图形碳(non-gC)原子的总数来衡量的。我们的计算表明,该参数会高估低氧/碳比时的缺陷密度。在此,我们采用缺陷的相对面积作为衡量缺陷密度的替代指标。由于该指标只针对缺陷,对结构的不规则性非常敏感,因此在缺陷密度较低和较高时都能很好地发挥作用。在另一个例子中,我们考虑了在不同的 O/C 比率下将 GO 还原成还原型氧化石墨烯(rGO)的情况,其中非 GC 原子数量的减少与相对较大缺陷的形成有关。因此,尚不清楚缺陷密度的减少程度(如果有的话)是否会随 O/C 比的变化而单调变化。缺陷面积与非气相化学原子数不同,它反映了这种模糊性,而且与 O/C 比的变化并非严格的单调变化。此外,我们还研究了缺陷分布和缺陷面积与模拟单元大小的关系。
Oxidation of graphite and subsequent exfoliation leads to single layer graphene oxide (GO). One of the key structural features of GO is the presence of different kinds of defects that dictates their various physical and chemical properties. Molecular dynamics simulations with ReaxFF force fields have been widely used to generate realistic models of GO. In these simulations, the extent and distribution of the defects are varied by changing the initial oxygen (O)/carbon (C) ratio while the defect density is often measured by the total number of non-graphitic carbon (non-gC) atoms. Our calculations suggest that this parameter overestimates the defect densities at low O/C ratio. Herein, we employ the relative area of the defects as an alternative metric to gauge the defect density. Being exclusive to the defects and sensitive to the structural irregularities, this metric works well at both low and high defect densities. In another example, we consider the reduction of GO to reduced graphene oxide (rGO) for different O/C ratios, where the decrease in the number of non-gC atoms is associated with the formation of comparatively larger defects. Hence, it is unclear whether the extent of reduction in the defect density (if at all) should vary monotonically with the O/C ratio. The defect area, unlike the count of the non-gC atoms, mirrors this ambiguity and the change with respect to O/C ratio is not strictly monotonic. Additionally, we also investigate the dependence of the defect distribution and defect area on the size of the simulation cell.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


