利用深度学习技术对全基因组和复杂基因组区域进行基因型推算的方法。
IF 2.6
3区 生物学
Q2 GENETICS & HEREDITY
引用次数: 0
摘要
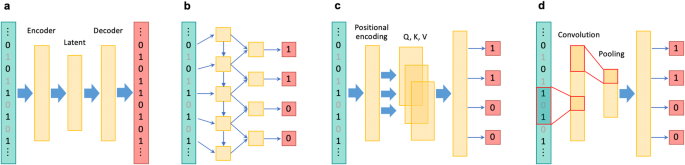
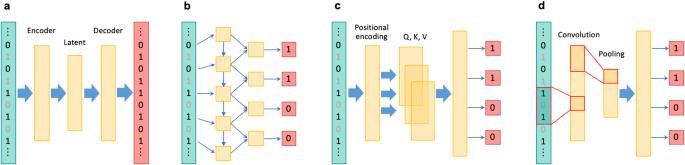
在人类遗传学研究中,尤其是在提高全基因组关联研究的能力和进行后续精细图谱绘制时,对未测量的基因型进行估算至关重要。最近,针对全基因组变异开发出了几种基于深度学习的基因型估算方法,这些方法具有学习复杂连锁不平衡模式的能力。此外,基于深度学习的估算方法还被应用于一个独特的基因组区域,即主要组织相容性复合体,被称为 HLA 估算。尽管目前基于深度学习的基因型估算方法具有各种优势,但它们也有一定的局限性,尚未成为标准方法。这些局限性包括与统计方法和传统的基于机器学习的方法相比,准确率的提高幅度不大。然而,这些方法的优点还包括其他方面,如 "无参照 "的特性可确保完全的隐私保护,以及更高的计算效率。此外,随着深度学习技术的不断发展,未来有望进一步提高预测准确性和可用性。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Genotype imputation methods for whole and complex genomic regions utilizing deep learning technology
The imputation of unmeasured genotypes is essential in human genetic research, particularly in enhancing the power of genome-wide association studies and conducting subsequent fine-mapping. Recently, several deep learning-based genotype imputation methods for genome-wide variants with the capability of learning complex linkage disequilibrium patterns have been developed. Additionally, deep learning-based imputation has been applied to a distinct genomic region known as the major histocompatibility complex, referred to as HLA imputation. Despite their various advantages, the current deep learning-based genotype imputation methods do have certain limitations and have not yet become standard. These limitations include the modest accuracy improvement over statistical and conventional machine learning-based methods. However, their benefits include other aspects, such as their “reference-free” nature, which ensures complete privacy protection, and their higher computational efficiency. Furthermore, the continuing evolution of deep learning technologies is expected to contribute to further improvements in prediction accuracy and usability in the future.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Journal of Human Genetics
生物-遗传学
CiteScore
7.20
自引率
0.00%
发文量
101
审稿时长
4-8 weeks
期刊介绍:
The Journal of Human Genetics is an international journal publishing articles on human genetics, including medical genetics and human genome analysis. It covers all aspects of human genetics, including molecular genetics, clinical genetics, behavioral genetics, immunogenetics, pharmacogenomics, population genetics, functional genomics, epigenetics, genetic counseling and gene therapy.
Articles on the following areas are especially welcome: genetic factors of monogenic and complex disorders, genome-wide association studies, genetic epidemiology, cancer genetics, personal genomics, genotype-phenotype relationships and genome diversity.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


