Qingfei Song, Xingyu Zhang, Zekai Miao and Qingyong Meng*,
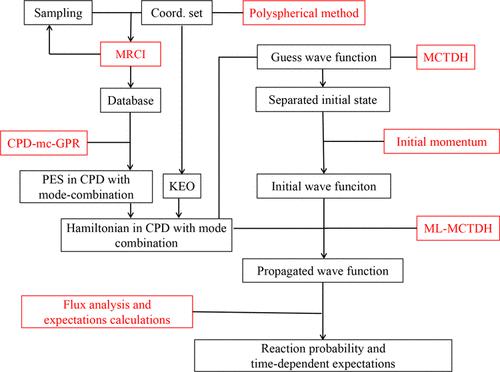
{"title":"为 OH + HO2 → O2 + H2O 的量子动力学构建基于网格表示法的模式组合哈密顿。","authors":"Qingfei Song, Xingyu Zhang, Zekai Miao and Qingyong Meng*, ","doi":"10.1021/acs.jctc.3c01090","DOIUrl":null,"url":null,"abstract":"<p >In this work, a systematic construction framework on a mode-combination Hamiltonian operator of a typical polyatomic reaction, OH + HO<sub>2</sub> → O<sub>2</sub> + H<sub>2</sub>O, is developed. First, a set of Jacobi coordinates are employed to construct the kinetic energy operator (KEO) through the polyspherical approach ( <cite><i>Phys. Rep.</i></cite> <span>2009</span>, <em>484</em>, 169). Second, due to the multiconfigurational electronic structure of this system, a non-adiabatic potential energy surface (PES) is constructed where the first singlet and triplet states are involved with spin–orbital coupling. To improve the training database, the training set of random energy data was optimized through a popular iterative optimization approach with extensive trajectories. Here, we propose an automatic trajectory method, instead of the classical trajectory on a crude PES, where the gradients are directly computed by the present <i>ab initio</i> calculations. Third, on the basis of the training set, the potential function is directly constructed in the canonical polyadic decomposition (CPD) form ( <cite><i>J. Chem. Theory Comput.</i></cite> <span>2021</span>, <em>17</em>, 2702−2713) which is helpful in propagating the nuclear wave function under the grid-based representation. To do this, the Gaussian process regression (GPR) approach for building the CPD form, called the CPD-GPR method ( <cite><i>J. Phys. Chem. Lett.</i></cite> <span>2022</span>, <em>13</em>, 11128−11135) is adopted where we further revise CPD-GPR by introducing the mode-combination (mc) scheme leading to the present CPD-mc-GPR approach. Constructing the full-dimension non-adiabatic Hamiltonian operator with mode combination, as test calculations, the nuclear wave function is propagated to preliminarily compute the reactive probability of OH + HO<sub>2</sub> → O<sub>2</sub> + H<sub>2</sub>O where the reactants are prepared in vibrational ground states and in the first triplet electronic state.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"20 2","pages":"597–613"},"PeriodicalIF":5.5000,"publicationDate":"2024-01-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Construction of a Mode-Combination Hamiltonian under the Grid-Based Representation for the Quantum Dynamics of OH + HO2 → O2 + H2O\",\"authors\":\"Qingfei Song, Xingyu Zhang, Zekai Miao and Qingyong Meng*, \",\"doi\":\"10.1021/acs.jctc.3c01090\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >In this work, a systematic construction framework on a mode-combination Hamiltonian operator of a typical polyatomic reaction, OH + HO<sub>2</sub> → O<sub>2</sub> + H<sub>2</sub>O, is developed. First, a set of Jacobi coordinates are employed to construct the kinetic energy operator (KEO) through the polyspherical approach ( <cite><i>Phys. Rep.</i></cite> <span>2009</span>, <em>484</em>, 169). Second, due to the multiconfigurational electronic structure of this system, a non-adiabatic potential energy surface (PES) is constructed where the first singlet and triplet states are involved with spin–orbital coupling. To improve the training database, the training set of random energy data was optimized through a popular iterative optimization approach with extensive trajectories. Here, we propose an automatic trajectory method, instead of the classical trajectory on a crude PES, where the gradients are directly computed by the present <i>ab initio</i> calculations. Third, on the basis of the training set, the potential function is directly constructed in the canonical polyadic decomposition (CPD) form ( <cite><i>J. Chem. Theory Comput.</i></cite> <span>2021</span>, <em>17</em>, 2702−2713) which is helpful in propagating the nuclear wave function under the grid-based representation. To do this, the Gaussian process regression (GPR) approach for building the CPD form, called the CPD-GPR method ( <cite><i>J. Phys. Chem. Lett.</i></cite> <span>2022</span>, <em>13</em>, 11128−11135) is adopted where we further revise CPD-GPR by introducing the mode-combination (mc) scheme leading to the present CPD-mc-GPR approach. Constructing the full-dimension non-adiabatic Hamiltonian operator with mode combination, as test calculations, the nuclear wave function is propagated to preliminarily compute the reactive probability of OH + HO<sub>2</sub> → O<sub>2</sub> + H<sub>2</sub>O where the reactants are prepared in vibrational ground states and in the first triplet electronic state.</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\"20 2\",\"pages\":\"597–613\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2024-01-10\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jctc.3c01090\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.3c01090","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Construction of a Mode-Combination Hamiltonian under the Grid-Based Representation for the Quantum Dynamics of OH + HO2 → O2 + H2O
In this work, a systematic construction framework on a mode-combination Hamiltonian operator of a typical polyatomic reaction, OH + HO2 → O2 + H2O, is developed. First, a set of Jacobi coordinates are employed to construct the kinetic energy operator (KEO) through the polyspherical approach ( Phys. Rep.2009, 484, 169). Second, due to the multiconfigurational electronic structure of this system, a non-adiabatic potential energy surface (PES) is constructed where the first singlet and triplet states are involved with spin–orbital coupling. To improve the training database, the training set of random energy data was optimized through a popular iterative optimization approach with extensive trajectories. Here, we propose an automatic trajectory method, instead of the classical trajectory on a crude PES, where the gradients are directly computed by the present ab initio calculations. Third, on the basis of the training set, the potential function is directly constructed in the canonical polyadic decomposition (CPD) form ( J. Chem. Theory Comput.2021, 17, 2702−2713) which is helpful in propagating the nuclear wave function under the grid-based representation. To do this, the Gaussian process regression (GPR) approach for building the CPD form, called the CPD-GPR method ( J. Phys. Chem. Lett.2022, 13, 11128−11135) is adopted where we further revise CPD-GPR by introducing the mode-combination (mc) scheme leading to the present CPD-mc-GPR approach. Constructing the full-dimension non-adiabatic Hamiltonian operator with mode combination, as test calculations, the nuclear wave function is propagated to preliminarily compute the reactive probability of OH + HO2 → O2 + H2O where the reactants are prepared in vibrational ground states and in the first triplet electronic state.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


