{"title":"高效捕获方法,用于识别各种遗传性视网膜疾病。","authors":"Hsiao-Jung Kao, Ting-Yi Lin, Feng-Jen Hsieh, Jia-Ying Chien, Erh-Chan Yeh, Wan-Jia Lin, Yi-Hua Chen, Kai-Hsuan Ding, Yu Yang, Sheng-Chu Chi, Ping-Hsing Tsai, Chih-Chien Hsu, De-Kuang Hwang, Hsien-Yang Tsai, Mei-Ling Peng, Shi-Huang Lee, Siu-Fung Chau, Chen Yu Chen, Wai-Man Cheang, Shih-Jen Chen, Pui-Yan Kwok, Shih-Hwa Chiou, Mei-Yeh Jade Lu, Shun-Ping Huang","doi":"10.1038/s41525-023-00388-3","DOIUrl":null,"url":null,"abstract":"<p><p>Our study presents a 319-gene panel targeting inherited retinal dystrophy (IRD) genes. Through a multi-center retrospective cohort study, we validated the assay's effectiveness and clinical utility and characterized the mutation spectrum of Taiwanese IRD patients. Between January 2018 and May 2022, 493 patients in 425 unrelated families, all initially suspected of having IRD without prior genetic diagnoses, underwent detailed ophthalmic and physical examinations (with extra-ocular features recorded) and genetic testing with our customized panel. Disease-causing variants were identified by segregation analysis and clinical interpretation, with validation via Sanger sequencing. We achieved a read depth of >200× for 94.2% of the targeted 1.2 Mb region. 68.5% (291/425) of the probands received molecular diagnoses, with 53.9% (229/425) resolved cases. Retinitis pigmentosa (RP) is the most prevalent initial clinical impression (64.2%), and 90.8% of the cohort have the five most prevalent phenotypes (RP, cone-rod syndrome, Usher's syndrome, Leber's congenital amaurosis, Bietti crystalline dystrophy). The most commonly mutated genes of probands that received molecular diagnosis are USH2A (13.7% of the cohort), EYS (11.3%), CYP4V2 (4.8%), ABCA4 (4.5%), RPGR (3.4%), and RP1 (3.1%), collectively accounted for 40.8% of diagnoses. We identify 87 unique unreported variants previously not associated with IRD and refine clinical diagnoses for 21 patients (7.22% of positive cases). We developed a customized gene panel and tested it on the largest Taiwanese cohort, showing that it provides excellent coverage for diverse IRD phenotypes.</p>","PeriodicalId":19273,"journal":{"name":"NPJ Genomic Medicine","volume":"9 1","pages":"4"},"PeriodicalIF":4.7000,"publicationDate":"2024-01-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10776681/pdf/","citationCount":"0","resultStr":"{\"title\":\"Highly efficient capture approach for the identification of diverse inherited retinal disorders.\",\"authors\":\"Hsiao-Jung Kao, Ting-Yi Lin, Feng-Jen Hsieh, Jia-Ying Chien, Erh-Chan Yeh, Wan-Jia Lin, Yi-Hua Chen, Kai-Hsuan Ding, Yu Yang, Sheng-Chu Chi, Ping-Hsing Tsai, Chih-Chien Hsu, De-Kuang Hwang, Hsien-Yang Tsai, Mei-Ling Peng, Shi-Huang Lee, Siu-Fung Chau, Chen Yu Chen, Wai-Man Cheang, Shih-Jen Chen, Pui-Yan Kwok, Shih-Hwa Chiou, Mei-Yeh Jade Lu, Shun-Ping Huang\",\"doi\":\"10.1038/s41525-023-00388-3\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Our study presents a 319-gene panel targeting inherited retinal dystrophy (IRD) genes. Through a multi-center retrospective cohort study, we validated the assay's effectiveness and clinical utility and characterized the mutation spectrum of Taiwanese IRD patients. Between January 2018 and May 2022, 493 patients in 425 unrelated families, all initially suspected of having IRD without prior genetic diagnoses, underwent detailed ophthalmic and physical examinations (with extra-ocular features recorded) and genetic testing with our customized panel. Disease-causing variants were identified by segregation analysis and clinical interpretation, with validation via Sanger sequencing. We achieved a read depth of >200× for 94.2% of the targeted 1.2 Mb region. 68.5% (291/425) of the probands received molecular diagnoses, with 53.9% (229/425) resolved cases. Retinitis pigmentosa (RP) is the most prevalent initial clinical impression (64.2%), and 90.8% of the cohort have the five most prevalent phenotypes (RP, cone-rod syndrome, Usher's syndrome, Leber's congenital amaurosis, Bietti crystalline dystrophy). The most commonly mutated genes of probands that received molecular diagnosis are USH2A (13.7% of the cohort), EYS (11.3%), CYP4V2 (4.8%), ABCA4 (4.5%), RPGR (3.4%), and RP1 (3.1%), collectively accounted for 40.8% of diagnoses. We identify 87 unique unreported variants previously not associated with IRD and refine clinical diagnoses for 21 patients (7.22% of positive cases). We developed a customized gene panel and tested it on the largest Taiwanese cohort, showing that it provides excellent coverage for diverse IRD phenotypes.</p>\",\"PeriodicalId\":19273,\"journal\":{\"name\":\"NPJ Genomic Medicine\",\"volume\":\"9 1\",\"pages\":\"4\"},\"PeriodicalIF\":4.7000,\"publicationDate\":\"2024-01-09\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10776681/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"NPJ Genomic Medicine\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1038/s41525-023-00388-3\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"NPJ Genomic Medicine","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1038/s41525-023-00388-3","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Highly efficient capture approach for the identification of diverse inherited retinal disorders.
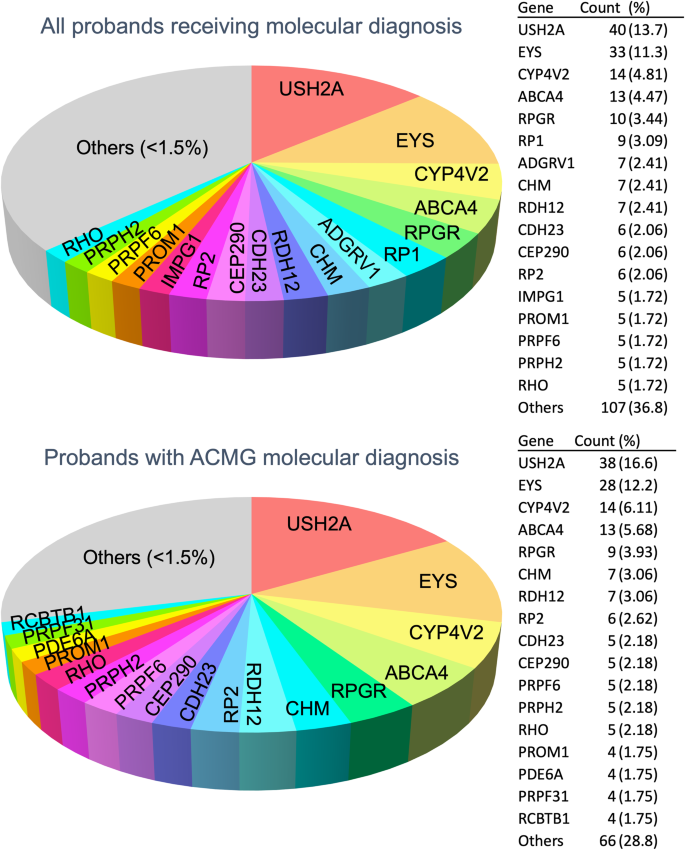
Our study presents a 319-gene panel targeting inherited retinal dystrophy (IRD) genes. Through a multi-center retrospective cohort study, we validated the assay's effectiveness and clinical utility and characterized the mutation spectrum of Taiwanese IRD patients. Between January 2018 and May 2022, 493 patients in 425 unrelated families, all initially suspected of having IRD without prior genetic diagnoses, underwent detailed ophthalmic and physical examinations (with extra-ocular features recorded) and genetic testing with our customized panel. Disease-causing variants were identified by segregation analysis and clinical interpretation, with validation via Sanger sequencing. We achieved a read depth of >200× for 94.2% of the targeted 1.2 Mb region. 68.5% (291/425) of the probands received molecular diagnoses, with 53.9% (229/425) resolved cases. Retinitis pigmentosa (RP) is the most prevalent initial clinical impression (64.2%), and 90.8% of the cohort have the five most prevalent phenotypes (RP, cone-rod syndrome, Usher's syndrome, Leber's congenital amaurosis, Bietti crystalline dystrophy). The most commonly mutated genes of probands that received molecular diagnosis are USH2A (13.7% of the cohort), EYS (11.3%), CYP4V2 (4.8%), ABCA4 (4.5%), RPGR (3.4%), and RP1 (3.1%), collectively accounted for 40.8% of diagnoses. We identify 87 unique unreported variants previously not associated with IRD and refine clinical diagnoses for 21 patients (7.22% of positive cases). We developed a customized gene panel and tested it on the largest Taiwanese cohort, showing that it provides excellent coverage for diverse IRD phenotypes.
NPJ Genomic MedicineBiochemistry, Genetics and Molecular Biology-Molecular Biology
CiteScore
9.40
自引率
1.90%
发文量
67
审稿时长
17 weeks
期刊介绍:
npj Genomic Medicine is an international, peer-reviewed journal dedicated to publishing the most important scientific advances in all aspects of genomics and its application in the practice of medicine.
The journal defines genomic medicine as "diagnosis, prognosis, prevention and/or treatment of disease and disorders of the mind and body, using approaches informed or enabled by knowledge of the genome and the molecules it encodes." Relevant and high-impact papers that encompass studies of individuals, families, or populations are considered for publication. An emphasis will include coupling detailed phenotype and genome sequencing information, both enabled by new technologies and informatics, to delineate the underlying aetiology of disease. Clinical recommendations and/or guidelines of how that data should be used in the clinical management of those patients in the study, and others, are also encouraged.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


